DOI:
10.1039/C8RA06103C
(Paper)
RSC Adv., 2019,
9, 668-674
Role of the disulfide bond on the structure and activity of μ-conotoxin PIIIA in the inhibition of NaV1.4†
Received
19th July 2018
, Accepted 23rd October 2018
First published on 3rd January 2019
Abstract
μ-Conotoxin PIIIA, a peptide toxin isolated from Conus purpurascens, blocks the skeletal muscle voltage-gated sodium channel NaV1.4 with significant potency. PIIIA has three disulfide bonds, which contribute largely to its highly constrained and stable structure. In this study, a combination of experimental studies and computational modeling were performed to assess the effects of deletion of the disulfide bonds on the structure and activity of PIIIA. The final results indicate that the three disulfide bonds of PIIIA are required to produce the effective inhibition of NaV1.4, and the removal of any one of the disulfide bonds significantly reduces its binding affinity owing to secondary structure variation, among which the Cys11–Cys22 is the most important for sustaining the structure and activity of PIIIA.
1. Introduction
Voltage-gated sodium channels (VGSCs) are responsible for the initiation and propagation of action potentials in many organs from the nervous system to the heart and muscles.1–3 μ-Conotoxins, members of the M superfamily,4,5 contain 16 to 25 residues with six Cys residues arranged in a class III framework (–C1C2–C3–C4–C5C6–)6 and are considered to be specific antagonists of voltage-gated Na+ channels.7 The μ-conotoxin PIIIA was isolated from Conus purpurascens, an eastern Pacific fish hunting species of cone snails.8 It is a 22-amino acid peptide, consisting of six cysteines and forming three pairs of disulfide bonds.9 The μ-conotoxins PIIIA and GIIIA are both high-affinity antagonists with skeletal muscle Na channel subtypes from a variety of vertebrate system, and PIIIA is also an irreversible inhibitor in the amphibian system.10 PIIIA selectively inhibits the skeletal muscle channel NaV1.4 with a half-maximal inhibitory concentration (IC50) value in the nanomolar range (36 nM),8 and can also block rNaV1.2 and rNaV1.7 at IC50 values of 690 nM and 6.2 μm, respectively (Fig. 1).11 Hence, it is valuable for use as a probe to enable different subtypes to be distinguished when used in conjunction with other selective toxins.11,12 In addition, PIIIA can also inhibit the KV1.1 and KV1.6 channels in the nanomolar range.13
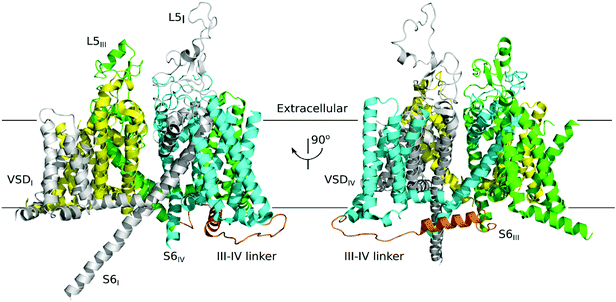 |
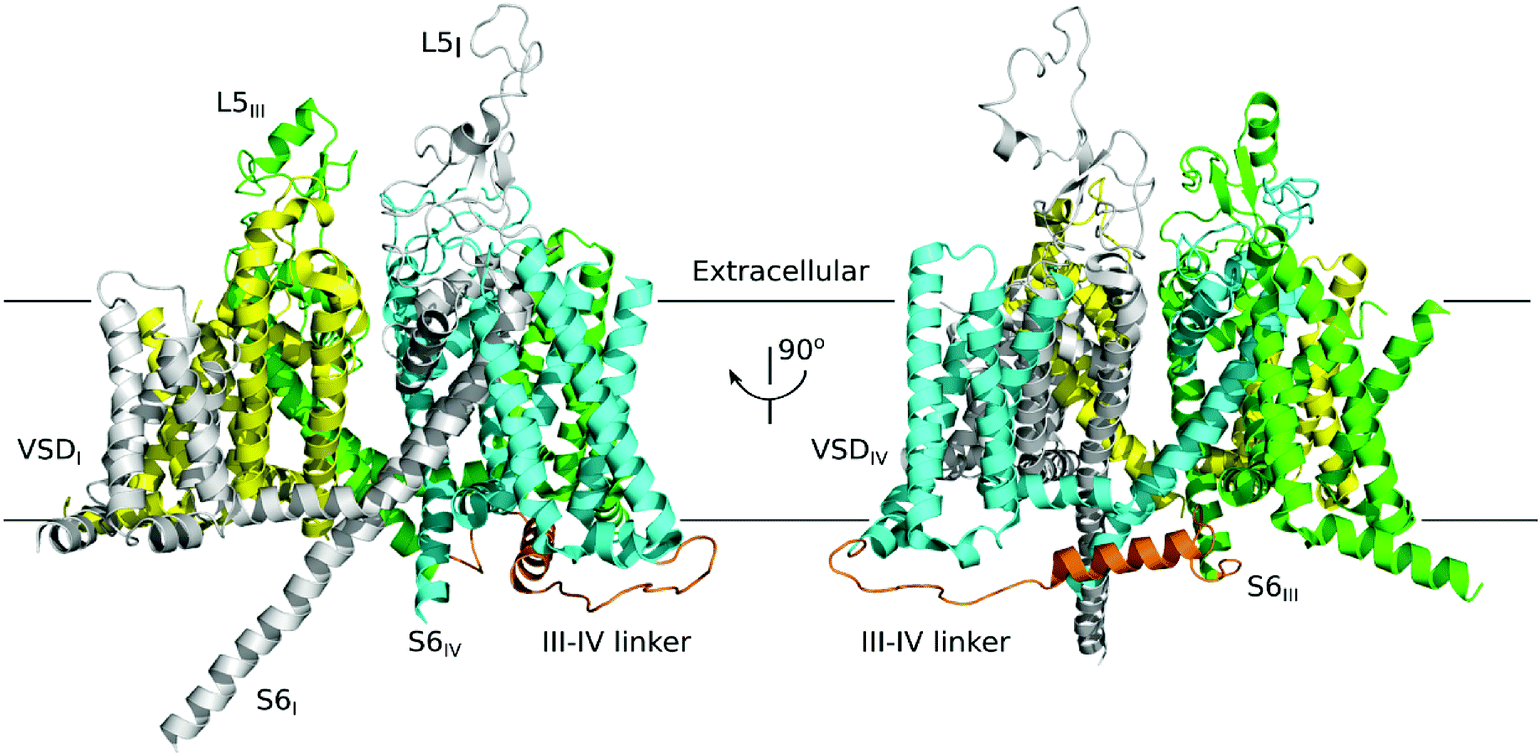
| | Fig. 1 Electron microscopy structure of the NaV1.4 of eukaryotic (PDB ID: 5XSY). The NaV α subunits have the same topological structure, and the single polypeptide chain folds into four repeats represented in blue, gray, yellow and green, each containing six transmembrane segments (TMs) (S1–S6). This NaV1.4 consists of the extracellular domain and the VSD (voltage sensing domain). | |
Related studies have shown that the disulfide connectivity alters the global fold of a toxin.14 Disulfide bonds in the peptide sequence give rise to a well-defined and constrained framework and generally play an important role in their biological action,15 but this also present challenges. The most critical step for the preparation of disulfide-bridged conopeptides is the oxidation to the correctly folded isomer16 and a related study has shown that the disulfide-bridges are susceptible to reduction in certain extracellular environments such as the blood.17 Thus, simplifying the disulfide bonds in peptides is of paramount importance for the application of peptides in pharmacy and other fields. Indeed, growing evidence suggests that many disulfide-rich peptides do not require all their native disulfide bonds to retain their biological activity.18,19 For instance, removal of the Cys1–Cys16 bridge in ω-conotoxin MVIIA does not abolish its calcium channel binding affinity.20 Some results have also shown that the key residues of KIIIA for voltage-gated sodium channel (VGSC) binding lie mostly on the alpha-helix and that the first disulfide bond can be removed without significantly affecting the structure of this helix, and its biological activity.21 This is not only isolate to the native peptide, removal of one of the disulfide bonds in the backbone cyclized Vc1.1 (cVc1.1) produced hcVc1.1, which maintained the secondary structure of the cVc1.1 and showed only a threefold decrease in the activity.22 Thus, it is possible to maintain the structure and activity of the peptide by the deletion of one of the disulfide bonds.
In this study, the contribution for each of the disulfide bonds to the structure and activity of the PIIIA was investigated by a combination of experimental studies and computational modeling. The produced results could be used as guidance for further engineering of the structure of PIIIA in future studies.
2. Materials and methods
2.1 Chemical synthesis of the peptides
The peptides were synthesized using a 9-fluorenylmethoxycarbonyl (Fmoc) solid phase peptide synthesis (SPPS) as described previously.23 The substitution of pyroglutamic with alanine does not affect the inhibitory activity as illustrated by the fact that the pyroglutamic acid plays a minor role in the binding between the PIIIA and NaV1.4.24 Thus, it is feasible to replace the pyroglutamic acid with another amino acid. In consideration of the structural similarity between pyroglutamic acid and proline, proline was used to replace the pyroglutamic acid to give minimal structure perturbation. The four cysteine residues in the disulfide deleted analogues were protected by using trityl and Acm groups for the selective oxidation. A two-step oxidation protocol was used to selectively fold the peptides. The first disulfide bond was oxidized in 0.1 M NH4HCO3 (pH 8–8.5) at a peptide concentration of 0.3 mg mL−1 with stirring for 24 h at room temperature. Afterward, the products were purified using reversed-phase high performance liquid chromatography (RP-HPLC) on a Phenomenex C18 column (250 × 10 mm, 10 μm).
Formation of the second disulfide bond was performed by dissolving 10 mg peptide in 10 mL (c = 0.42 mM) buffer A/B (Table S1†), and adding 0.2 mL I2 solution dropwise (5 mg I2 dissolved in 1 mL CH3CN) with stirring for 30 min, then quenching with ascorbic acid (5 mg ascorbic acid dissolved in 1 mL H2O). After the oxidation, the products were purified using RP-HPLC. After confirming the product peak using mass spectrometry, the solid peptide was obtained by lyophilization. Analytical RP-HPLC with a gradient of 0–80% buffer B (the specific concentration ratio is shown in Table S1†) and a flow rate of 1 mL min−1 in 40 min and electrospray-mass spectroscopy was used to confirm the purity and molecular mass of the synthesized peptides (Fig. S1 and Table S4†).
2.2 Circular dichroism spectra
The circular dichroism (CD) spectra of PIIIA* and its analogues were performed on a Jasco J-810 spectropolarimeter over the wavelength range of 250–190 nm using a 1.0 mm path length cell, a bandwidth of 1.0 nm, a response time of 2 s, and the results were averaged over three scans. The spectra were recorded at room temperature under a nitrogen atmosphere. A blank scan was performed before the analysis of the samples was performed. The peptides were dissolved with buffer A or B (Table S1†), reaching a concentration of 0.1 mmol L−1. The quality of each peptide can be found in Table S2.† The molar ellipticity [θ] were fitted with linear regression using the following equation:
In which mdeg represents the raw CD data, C represents the peptide molar concentration (mM), and l represents the cell path length (mm).
2.3 Electrophysiology
Whole-cell patch-clamp recordings from HEK293 cells were performed using an Axon digidata 1550A amplifier. The α subunit of the NaV1.4 oocytes was prepared and a double electrode voltage clamp was used for recording. Similar to previous studies,25–31 the cells were perfused with extracellular solution containing 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose. The pH of the solution was adjusted to 7.4 using NaOH. The oocytes were placed in a vial containing 30 μL ND96 and two electrodes voltage clamped in the −80 mV holding potential chamber, repeating every 10 seconds, each time with 10 millisecond square wave membrane potential depolarized to −10 mV, to activate the NaV1.4 ion channel current. The maximum current caused by the voltage-clamp square wave was detected and the compound was tested by perfusion after being stabilized. The compounds were dissolved in extracellular fluid and perfused using a self-gravity perfusion system. Extracellular fluid containing 130 mM Cs aspartate, 5 mM MgCl2, 5 mM EGTA, 10 mM HEPES and 4 mM Tris-ATP was used with the pH adjusted to 7.2 by addition of CsOH. The test concentrations of the peptides were set at 0.1, 0.3, 1, 1, 3, and 10 μm (Table S3†), respectively. At least two cells were tested at each concentration. After the current caused by the voltage clamp square wave was stabilized, the changes in the current size before and after the use of the peptide were compared and the blocking effects of the peptide on the NaV1.4 channels was calculated.
2.4 Homology modeling
The NMR structure of PIIIA (PDB code: 1R9I) was obtained from the protein data bank (http://www.rcsb.org/). PIIIA* and its three analogues, each with one different pair of disulfide-bonded cysteine residues replaced with alanine, were constructed as shown in Table 1. The three disulfide bonds (Cys4–Cys16, Cys5–Cys21, and Cys11–Cys22) of PIIIA* were numbered as 1, 2 and 3, respectively, and the corresponding disulfide deleted analogues were named as PIIIA*-x (x = 1, 2, 3), in which each number represents that the corresponding disulfide bond that was removed. PIIIA*-1, PIIIA*-2, and PIIIA*-3 were constructed in MODELLER (version 9v16),32,33 as described previously.34 PIIIA* and its analogues was subjected to further refinement of the molecular dynamics (MD) simulations.
Table 1 Sequence of μ-conotoxins PIIIA and its analoguesa
| Peptide |
Name |
Sequence |
| PIIIA* is an analogue of PIIIA in which the first amino acid is replaced using Pro. PIIIA*-1, 2, 3 represents the analogue missing the first (Cys4–Cys16), second (Cys5–Cys21) and third (Cys11–Cys22) disulfide bond, respectively. |
| PIIIA |
PIIIA |
 |
| PIIIA* |
PIIIA* |
 |
| PIIIA*-1 |
PIIIA*[C4A,C16A] |
 |
| PIIIA*-2 |
PIIIA*[C5A,C21A] |
 |
| PIIIA*-3 |
PIIIA*[C11A,C22A] |
 |
2.5 Molecular dynamics simulation
By modifying the model we built and validated previously,35 we obtained the complexes: NaV1.4/PIIIA*-1, NaV1.4/PIIIA*-2, and NaV1.4/PIIIA*-3. These complexes were subjected to MD simulations for refinement of the PIIIA* binding mode in AMBER 16.36 After building the whole system in LEaP, a module of AMBER16, minimization was performed to remove the van der Waals clashes between PIIIA* and NaV1.4. Other details regarding the methods used for MD simulation are described in our previous study.37 PIIIA* and its analogues were refined using 100 ns MD simulations, while the complexes were refined using 20 ns MD simulations. The last conformation was extracted from the MD trajectory.
2.6 Binding energy calculations using MMGB/SA
A molecular mechanics generalized Born surface area (MMGB/SA)38 was applied to calculate the energy contributed by the key amino acids of the peptide. For efficient conformation sampling, the binding energy calculation was performed on the five MD trajectories using MMGB/SA and the averaged values were used as the final result. The parameters used for setting up were the same as those previously described.34
3. Results and discussion
3.1 Chemical synthesis of peptides
After the solid phase reaction had finished, half of the resin peptide was partially deprotected and cleaved from the resin with TFA. For PIIIA*-1, the protecting group of Cys5 and Cys21 is trityl (trt), while the protecting group of Cys11 and Cys22 is acetamidomethyl (Acm). The trt protection group of Cys5 and Cys21 can be deprotected in a mixture of TFA/TIS/H2O (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5), whereas the Acm protection group can only be removed in I2 solvent. Thus, in the first round oxidation in NH4HCO3, only the disulfide bond Cys5–Cys21 was formed, whereas the second disulfide bond Cys11–Cys22 was formed during the oxidation using I2 solvent. The product of each step was obtained by purification using RP-HPLC. The final product was obtained by freeze-drying and was purified and validated using RP-HPLC and MS (Fig. S1†).
:5:5), whereas the Acm protection group can only be removed in I2 solvent. Thus, in the first round oxidation in NH4HCO3, only the disulfide bond Cys5–Cys21 was formed, whereas the second disulfide bond Cys11–Cys22 was formed during the oxidation using I2 solvent. The product of each step was obtained by purification using RP-HPLC. The final product was obtained by freeze-drying and was purified and validated using RP-HPLC and MS (Fig. S1†).
3.2 Structural changes of PIIIA and its analogues
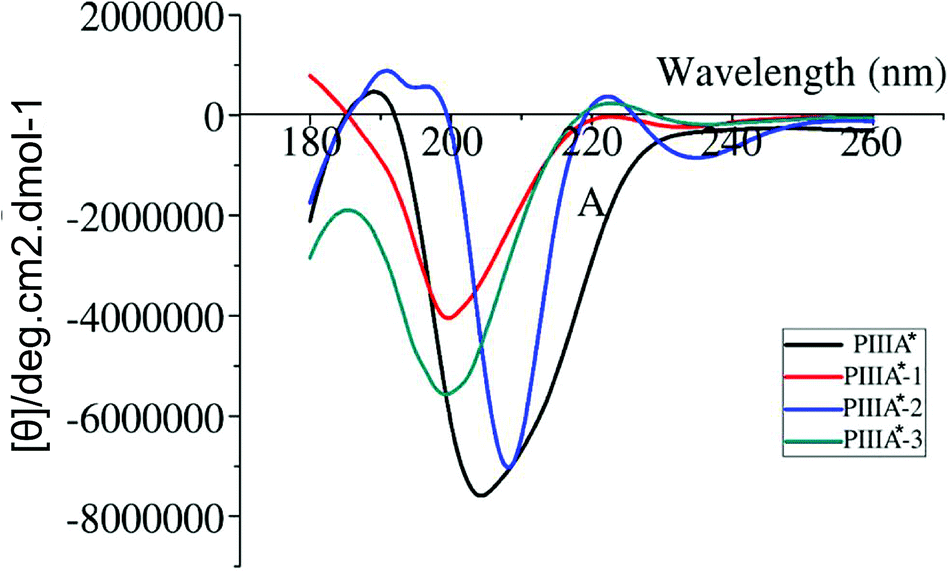
Deletion of the disulfide bond resulted in different CD spectra for PIIIA*-1, 2, and 3 (Fig. 2). In previous studies,39,40 an α-helix structure exhibited a strong negative band around 208 nm, and a strong positive band around 190 nm. It was observed from the CD spectra (Fig. 2) that PIIIA* has strong negative and positive bands close to 208 and 190 nm, respectively, indicating that PIIIA* contains the α-helix component. Similarly, PIIIA*-2 strongly absorbs at 209 and 190 nm, and thus it was deduced that it contain an α-helix secondary structure. In addition, PIIIA*-2 has a negative absorption around 198 nm and a small and wide positive absorption at 220 nm indicating that PIIIA*-2 also contains part of a random coil.39,40 In contrast, PIIIA*-1,3 only has a negative absorption at 198 nm and a small and wide positive absorption at 220 nm, indicating that most of the secondary structure of PIIIA*-1,3 is the random coil. Indeed, after the 100 ns MD simulation of PIIIA* and its analogues, we found that the structure of PIIIA*-1,3 contains a large number of random coils compared with PIIIA* (Fig. 3A and C). However, the structure of PIIIA*-2 is similar to that of PIIIA* and both contain an α-helix (Fig. 3B). This result is in line with our CD results. The α-helix in PIIIA*-2 enables a better stability than PIIIA*-1,3, which may lead to the inhibition activity of PIIIA*-2 being superior to that of PIIIA*-1,3. From the root mean square deviation (RMSD) analysis (Fig. 3D), the RMSD of PIIIA*-3 is greater than that of PIIIA*-1,2, suggesting that the conformation of PIIIA*-3 is the most significantly deviated from the initial conformation of the PIIIA* and its inhibitory activity is also the least active.
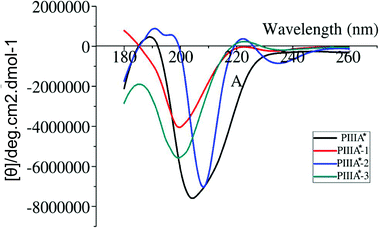 |
| | Fig. 2 The CD spectrum of PIIIA* and its analogues. The CD spectra of PIIIA*, PIIIA*-1, PIIIA*-2, and PIIIA*-3 are shown in black, red, blue and green, respectively. | |
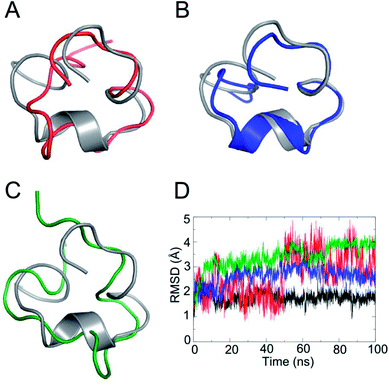 |
| | Fig. 3 Structure and stability comparison between PIIIA* and its disulfide deleted analogues. (A), (B), and (C) show the overlapped structures of PIIIA*-1, 2, and 3 with PIIIA*, respectively. Black represents PIIIA, and red, blue and green are PIIIA*-1, PIIIA*-2, and PIIIA*-3, respectively. (D) Shows an RMSD superposition diagram of PIIIA-1, 2, and 3. In (D) black, red, blue and green represent PIIIA*, PIIIA*-1, PIIIA*-2 and PIIIA*-3, respectively. The RMSD value was averaged for two MD trajectories. | |
3.3 Electrophysiology
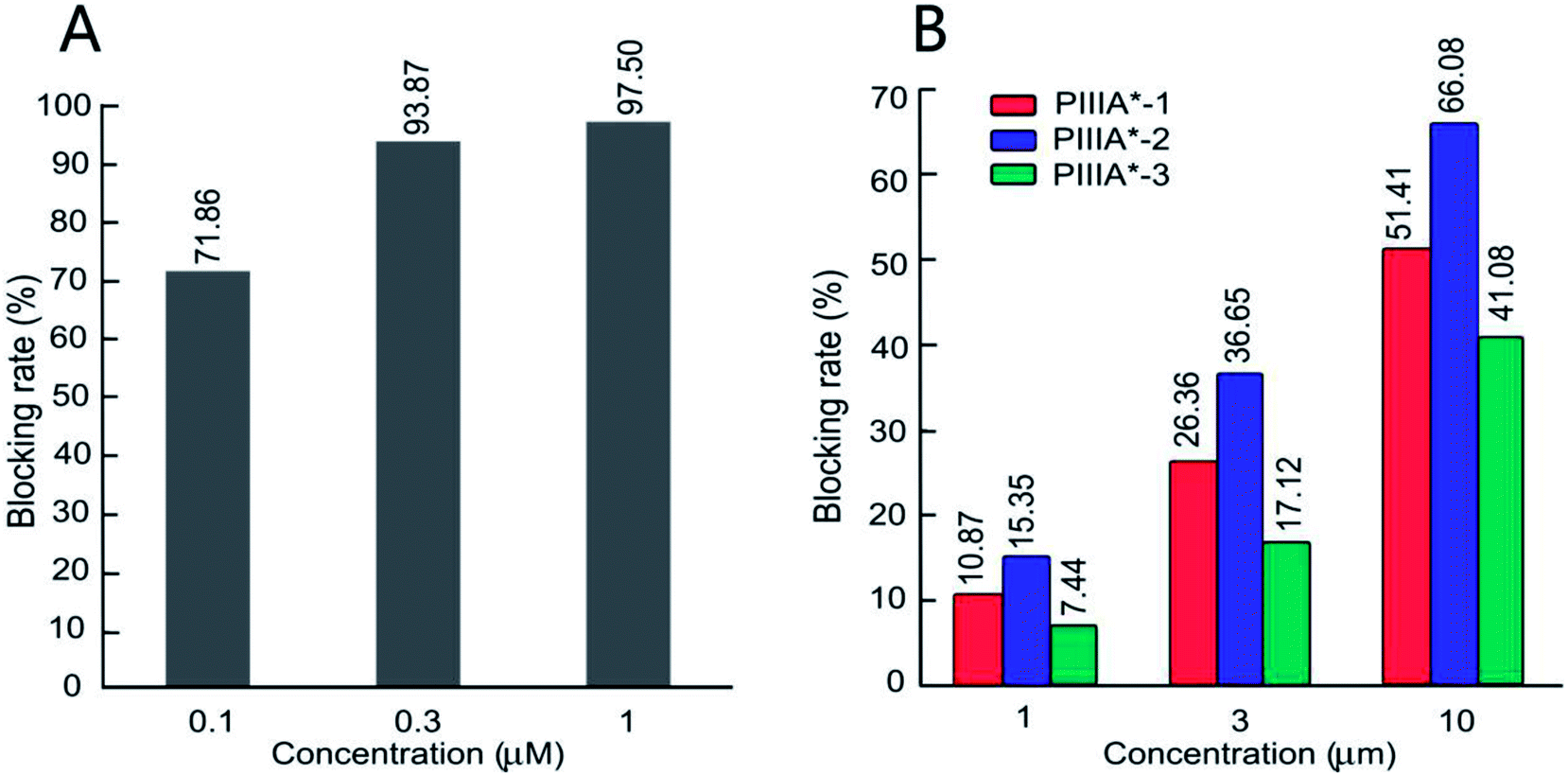
The biological activity of the four peptides (PIIIA*, PIIIA*-1, PIIIA*-2, and PIIIA*-3) on the hNaV1.4 channels were measured by conventional patch clamp protocols. Control experiments were performed on the PIIIA* blocking to hNaV1.4. Based on the percentage of blocking in a fixed peptide concentration, it was found that PIIIA*-2 had the best blocking effect on NaV1.4 at three different concentrations among the three disulfides deleted analogues, but is still less potent than PIIIA* (Fig. 4A and B). At a concentration of 10 μM (Fig. 4B), PIIIA* exhibits a much stronger blocking effect on the hNaV1.4 channel than PIIIA*-1, PIIIA*-2, and PIIIA*-3. The current blocking rates for PIIIA*-1, PIIIA*-2 and PIIIA*-3 are 51.41%, 66.08%, and 41.08%, respectively. These results suggest that deletion of any one of the disulfide bonds significantly decreases the activity of PIIIA*. The inhibitory activity of PIIIA*-2 is stronger than PIIIA*-1, while PIIIA*-3 has the worst inhibitory activity, indicating that the varied disulfide bonds play different roles on the activity of PIIIA*, with the disulfide bond Cys11–Cys22 contributing most to the activity, and the disulfide bond Cys5–Cys21 contributing least to the activity. Indeed, related studies also showed that all of the three disulfides, especially Cys3–Cys6, were important for the inhibition of NaV1.4 by GIIIA.41 Deletion of any pair of disulfide bond could have an influence on the structure of the PIIIA and the GIIIA, and thus affect their inhibitory activity on the sodium channels. For BuIIIB, however, the Cys1–Cys4 bridge was more critical for NaV1.3 blocking,42 which indicates that the disulfide bond plays a different role in these three conotoxins. Exploring the effects of the disulfide bonds on the structure of PIIIA is particularly important to the rational design of PIIIA analogues.
 |
| | Fig. 4 Inhibition activity of the PIIIA* disulfide deleted analogues at NaV1.4. (A) Represents the inhibition rate of PIIIA* on hNaV1.4 at different concentrations. (B) The inhibition rate of PIIIA*-1, 2, and 3 on hNaV1.4 at different concentrations. | |
3.4 Binding modes of NaV1.4 with the PIIIA analogues
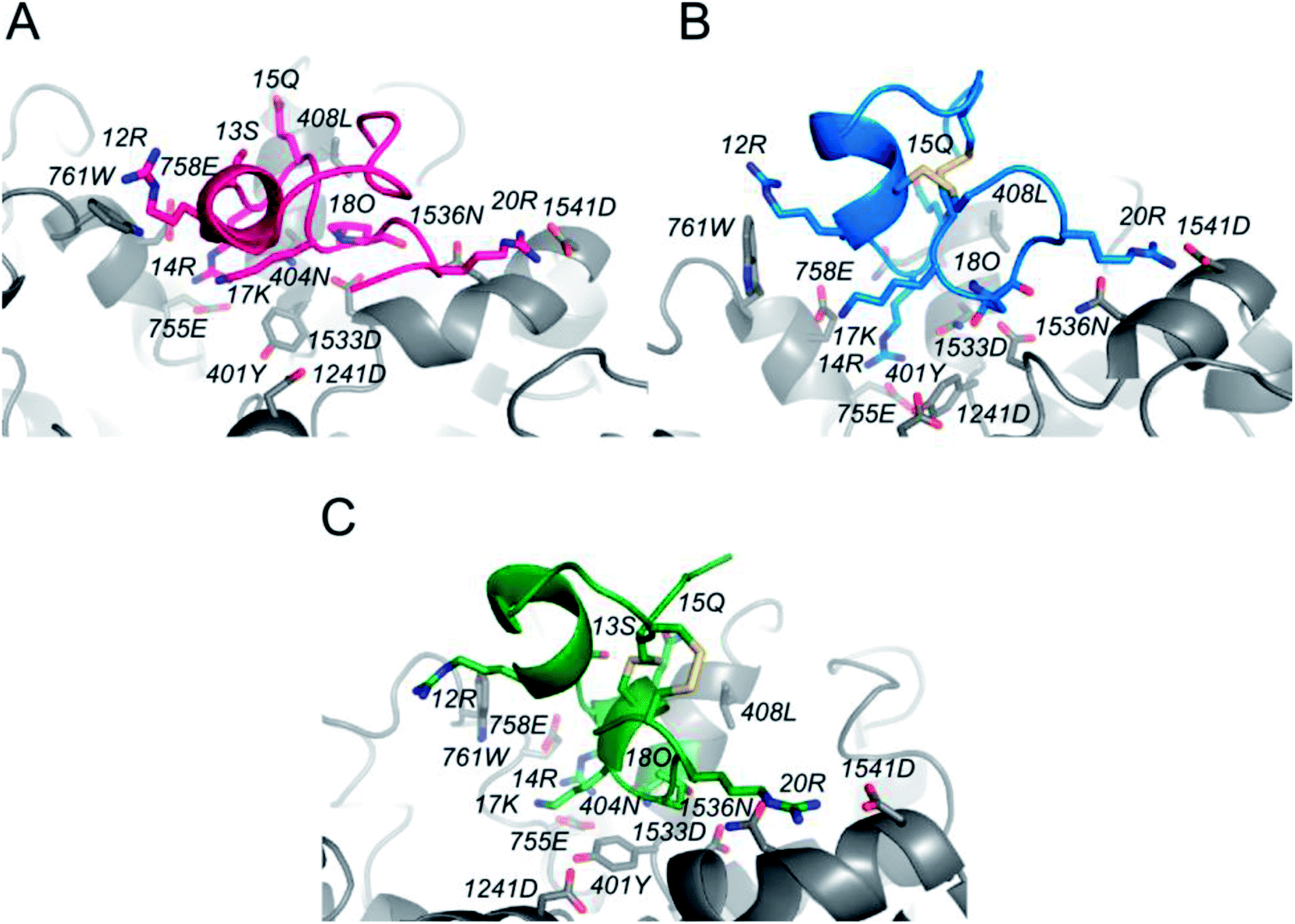
In order to determine the binding mode of PIIIA* at NaV1.4, a 20 ns MD simulation was applied on a NaV1.4/PIIIA*-1, 2, and 3 system, and each system was repeated five times. PIIIA is rich with positively charged residues, and is overall complementary to NaV1.4, leading to the formation of numerous salt-bridges at the interface between PIIIA and NaV1.4.35 Previous mutagenesis data indicates that R12, R14, and R20 contribute most to the binding affinity of PIIIA to NaV1.4.43 Disulfide bond deletion affects the secondary structure of PIIIA*-1, 2, and 3, weakening their interaction with NaV1.4. The R12 of PIIIA*-1, 2, and 3 only forms a very weak hydrogen bond interaction with W761, but the R12 of PIIIA forms salt-bridges with the E758 that is close to the pore filter.35 Owing to the migration of R14, the R14 of PIIIA*-1, 2, and 3 forms salt-bridges with E755 that are weaker than that of PIIIA. As suggested by the results from the aforementioned electrophysiological testing, removal of the disulfide bond results in a significantly decreased binding affinity. In comparison with PIIIA*-1 and PIIIA*-3, the R20 of PIIIA-2 forms salt-bridges with D1541 at the periphery of the binding site, but the R20 of PIIIA*-1,3 is deflected, resulting in a deviation from D1541 and weakening of the salt-bridges. Therefore, the inhibition activity of PIIIA*-2 is stronger than PIIIA*-1 and PIIIA*-3. The results from the RMSD analysis (Fig. S3†) suggest that the three complexes reached equilibrium after a 20 ns MD simulation, with a RMSD of less than 1.2 Å, indicating that the NaV1.4/PIIIA*-1, 2 and 3 systems were all relatively stable in MD.
3.5 Binding energy calculation and decomposition
In order to explore the effects of deletion of the disulfide bonds on the binding affinity of PIIIA* to the NaV1.4 channel, we used MMGB/SA to calculate the energy contribution provided by both of these key residues of the binding site (Table 3) and the residues on PIIIA*-1, 2, and 3 (Fig. S3†). Meanwhile, the interaction between these key amino acids and NaV1.4 were analyzed using the binding modes (Fig. 5). The binding energy calculations suggest that the binding energy of PIIIA*-2 to NaV1.4 is lower than PIIIA*-1, 3 but that they are all much lower than PIIIA* (Table 3), which is consistent with the previously mentioned electrophysiological testing. It is indicated that deletion of the disulfide bond resulted in a significantly decreased affinity of PIIIA*. Mutagenesis studies show that the major residues that affect the interaction between PIIIA and NaV1.4 channel are R12, R14, and R20.43 Analyzing the energy contribution of these key residues in PIIIA*-1, 2, and 3, we found that the binding energy contributed by R20 of PIIIA*-1, 2 is much lower than PIIIA*-1, 3. This explains why PIIIA*-2 is better in comparison to PIIIA*-1 and PIIIA*-3. Compared with the contributions to the binding free energy of R12 of PIIIA*-1, 2, 3 (−0.61 kcal mol−1, −0.49 kcal mol−1 and 0.02 kcal mol−1, respectively), the R12 of PIIIA* is much lower, about −3.54 kcal mol−1. In addition, the binding free energy of the R14 of PIIIA* is lower than PIIIA*-1, 2, and 3. This further confirms that deletion of the disulfide bond results in the significantly decreased affinity of PIIIA*. More detailed pairwise interactions between PIIIA and NaV1.4 are listed in Table 2.
Table 2 Interactions between the key amino acids on PIIIA-1, 2, 3 and NaV1.4
| Compounds |
R12 |
R14 |
Q15 |
K17 |
O18 |
R20 |
| PIIIA |
E758,W761 |
F401,N404 |
L408 |
D1241 |
N404,D1533 |
D1541 |
| PIIIA-1 |
W761 |
E755,E758 |
Q407 |
— |
N404,D1533,N1536 |
D1541 |
| PIIIA-2 |
W761 |
N404,E755 |
Q407 |
— |
N404,D1533,N1536 |
N1536,D1541 |
| PIIIA-3 |
— |
N404,E755,E758 |
Q407 |
— |
D1533,N1536 |
D1541 |
Table 3 The energy contribution of the key residues of the peptide
| Residues |
PIIIA*a (kcal mol−1) |
PIIIA*-1a (kcal mol−1) |
PIIIA*-2a (kcal mol−1) |
PIIIA*-3a (kcal mol−1) |
| The energy contribution was calculated using the MMGB/SA method and was averaged for five MD trajectories. |
| R12 |
−3.54(±0.966) |
−0.61(±0.122) |
−0.49(±0.244) |
0.02(±0.175) |
| R14 |
−15.16(±0.812) |
−11.83(±2.365) |
−10.88(±0.435) |
−12.94(±0.375) |
| R20 |
−7.46(±0.273) |
−6.27(±1.254) |
−8.46(±0.340) |
−3.84(±0.544) |
| Total energy (kcal mol−1) |
−51.06(±2.580) |
−23.30(±1.093) |
−25.02(±1.505) |
−20.25(±1.607) |
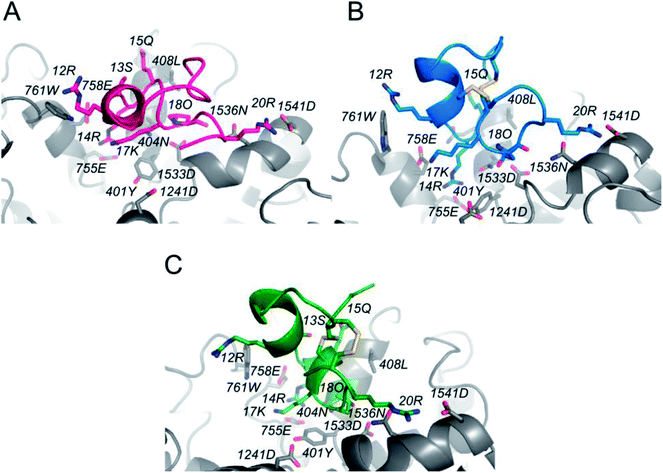 |
| | Fig. 5 The binding modes of NaV1.4 with PIIIA and its analogues. (A), (B) and (C) represent the combination pattern of NaV1.4/PIIIA*-1, NaV1.4/PIIIA*-2, and NaV1.4/PIIIA*-3, respectively. Red, blue and green represent PIIIA*-1, PIIIA*-2, and PIIIA*-3, respectively. | |
4. Conclusions and perspectives
The purpose of this experiment was to explore the contribution of three pairs of disulfide bonds on the structure and inhibitory activity of PIIIA* from the aspects of experimental and computational modeling studies. Using the method of homology modeling and MD simulations, the inhibitory activity of PIIIA*-1, 2, and 3 was found to be significantly lower than for PIIIA*, which is in accordance with the experimental results obtaining using electrophysiological studies. By calculation of the binding energy and decomposition, we found that the second disulfide bond (Cys5–Cys21) had the minimum influence on the structure and activity of NaV1.4, which is also in agreement with the results obtained from experimental studies. The blocking effects of PIIIA*-2 to NaV1.4 is in the order of PIIIA*-2 > PIIIA*-3 > PIIIA*-1. The binding energy of NaV1.4/PIIIA*-1, 2, and 3 also shows that the binding affinity of PIIIA*-2 on NaV1.4 is better compared to PIIIA*-1, and 3 on NaV1.4.
In conclusion, our study clearly shows that all three disulfide bonds in PIIIA are particularly important to maintaining the stability of the structure and to producing the effective inhibition of NaV1.4. Furthermore, the results indicate that the second disulfide bond (Cys5–Cys21) connection has the smallest influence on the structure and activity of the peptide with NaV1.4, followed by the first pair (Cys4–Cys6) and the third pair (Cys11–Cys22). The results from this study expand our knowledge of the importance of the three disulfide bonds in PIIIA, and will provide valuable information for the design of analogues with a better affinity and selectivity for therapeutic usage.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (No. 201762011 for R. Y.), the National Natural Science of China (NSFC 81561148012), a grant from the National Natural Science Foundation of China (NSFC) (No. 81502977 for R. Y.), the National Laboratory Director fund (QNLM201709), the Scientific and Technological Innovation Project Financially Supported by the Qingdao National Laboratory for Marine Science and Technology (No. 2015ASKJ02) and the Taishan Scholars Program of Shandong, China.
Notes and references
- H. Shen, Q. Zhou, X. Pan, Z. Li, J. Wu and N. Yan, Science, 2017, 355, eaal4326 CrossRef PubMed.
- M. Noda, T. Ikeda, H. Suzuki, H. Takeshima, T. Takahashi, M. Kuno and S. Numa, Nature, 1986, 322, 826–828 CrossRef CAS PubMed.
- B. Rudy, J. Physiol., 1978, 283, 1–21 CrossRef CAS.
- G. P. Corpuz, R. B. Jacobsen, E. C. Jimenez, M. Watkins, C. Walker, C. Colledge, J. E. Garrett, O. Mcdougal, W. Li and W. R. Gray, Biochemistry, 2005, 44, 8176 CrossRef CAS PubMed.
- R. B. Jacob and O. M. McDougal, Cell. Mol. Life Sci., 2010, 67, 17–27 CrossRef CAS PubMed.
- H. Terlau and B. M. Olivera, Physiol. Rev., 2004, 84, 41–68 CrossRef CAS PubMed.
- R. S. Norton, Molecules, 2010, 15, 2825–2844 CrossRef CAS PubMed.
- K. J. Shon, B. M. Olivera, M. Watkins, R. B. Jacobsen, W. R. Gray, C. Z. Floresca, L. J. Cruz, D. R. Hillyard, A. Brink, H. Terlau and D. Yoshikami, J. Neurosci., 1998, 18, 4473–4481 CrossRef CAS PubMed.
- A. A. Tietze, D. Tietze, O. Ohlenschläger, E. Leipold, F. Ullrich, T. Kühl, A. Mischo, G. Buntkowsky, M. Görlach, S. H. Heinemann and D. Imhof, Angew. Chem., Int. Ed. Engl., 2012, 51, 4058–4061 CrossRef CAS PubMed.
- K. J. Shon, B. M. Olivera, M. Watkins, R. B. Jacobsen, W. R. Gray, C. Z. Floresca, L. J. Cruz, D. R. Hillyard, A. Brink, H. Terlau and D. Yoshikami, J. Neurosci., 1998, 18, 4473–4481 CrossRef CAS.
- P. Safo, T. Rosenbaum, A. Shcherbatko, D. Y. Choi, E. Han, J. J. Toledo-Aral, B. M. Olivera, P. Brehm and G. Mandel, J. Neurosci., 2000, 20, 76–80 CrossRef CAS PubMed.
- N. R. Munasinghe and M. J. Christie, Toxins, 2015, 7, 5386–5407 CrossRef CAS PubMed.
- E. Leipold, F. Ullrich, M. Thiele, A. A. Tietze, H. Terlau, D. Imhof and S. H. Heinemann, Biochem. Biophys. Res. Commun., 2016, 482, 1135–1140 CrossRef PubMed.
- P. Heimer, A. A. Tietze, C. A. Bäuml, A. Resemann, F. J. Mayer, D. Suckau, O. Ohlenschläger, D. Tietze and D. Imhof, Anal. Chem., 2018, 90, 3321–3327 CrossRef CAS PubMed.
- M. Góngora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef PubMed.
- T. S. Kang, Z. Radić, T. T. Talley, S. D. S. Jois, P. Taylor and R. M. Kini, Biochemistry, 2007, 46, 3338–3355 CrossRef CAS PubMed.
- C. J. Armishaw, N. L. Daly, S. T. Nevin, D. J. Adams, D. J. Craik and P. F. Alewood, J. Biol. Chem., 2006, 281, 14136–14143 CrossRef CAS PubMed.
- P. Savarin, R. Romi-Lebrun, S. Zinn-Justin, B. Lebrun, T. Nakajima, B. Gilquin and A. Menez, Protein Sci., 1999, 8, 2672–2685 CrossRef CAS PubMed.
- Q. Zhu, S. Liang, L. Martin, S. Gasparini, A. Ménez and C. Vita, Biochemistry, 2002, 41, 11488–11494 CrossRef CAS PubMed.
- M. Price-Carter, M. S. Hull and D. P. Goldenberg, Biochemistry, 1998, 37, 9851–9861 CrossRef CAS PubMed.
- K. K. Khoo, Z.-P. Feng, B. J. Smith, M.-M. Zhang, D. Yoshikami, B. M. Olivera, G. Bulaj and R. S. Norton, Biochemistry, 2009, 48, 1210–1219 CrossRef CAS PubMed.
- R. Yu, S. N. Kompella, D. J. Adams, D. J. Craik and Q. Kaas, J. Med. Chem., 2013, 56, 3557–3567 CrossRef CAS PubMed.
- R. Yu, S. N. Kompella, D. J. Adams, D. J. Craik and Q. Kaas, J. Med. Chem., 2013, 56, 3557–3567 CrossRef CAS PubMed.
- K. J. Nielsen, M. Watson, D. J. Adams, A. K. Hammarström, P. W. Gage, J. M. Hill, D. J. Craik, L. Thomas, D. Adams, P. F. Alewood and R. J. Lewis, J. Biol. Chem., 2002, 277, 27247–27255 CrossRef CAS PubMed.
- Y. Chen, F. H. Yu, E. M. Sharp, D. Beacham, T. Scheuer and W. A. Catterall, Mol. Cell. Neurosci., 2008, 38, 607–615 CrossRef CAS PubMed.
- Y. Chen, F. H. Yu, D. J. Surmeier, T. Scheuer and W. A. Catterall, Neuron, 2006, 49, 409–420 CrossRef CAS PubMed.
- E. S. Bennett, J. Membr. Biol., 2004, 197, 155–168 CrossRef CAS PubMed.
- M. Chahine, P. B. Bennett, A. L. George and R. Horn, Pflügers Arch., 1994, 427, 136–142 CrossRef CAS PubMed.
- L. J. Cruz, W. R. Gray, B. M. Olivera, R. D. Zeikus, L. Kerr, D. Yoshikami and E. Moczydlowski, J. Biol. Chem., 1985, 260, 9280–9288 CAS.
- K. Schroeder, B. Neagle, D. J. Trezise and J. Worley, J. Biomol. Screening, 2003, 8, 50–64 CrossRef CAS PubMed.
- K. A. Young and J. H. Caldwell, J. Physiol., 2005, 565, 349–370 CrossRef CAS PubMed.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS PubMed.
- N. Eswar, B. John, N. Mirkovic, A. Fiser, V. A. Ilyin, U. Pieper, A. C. Stuart, M. A. Marti-Renom, M. S. Madhusudhan, B. Yerkovich and A. Sali, Nucleic Acids Res., 2003, 31, 3375–3380 CrossRef CAS PubMed.
- R. Yu, D. J. Craik and Q. Kaas, PLoS Comput. Biol., 2011, 7, e1002011 CrossRef CAS PubMed.
- F. Chen, W. Huang, T. Jiang and R. Yu, Mar. Drugs, 2018, 16(5), 153 CrossRef PubMed.
- D. A. Case, D. S. Cerutti, T. E. Cheatham, III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, D. Greene, N. Homeyer, S. Izadi, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2017, University of California, San Francisco, CA, 2017 Search PubMed.
- R. Yu, Q. Kaas and D. J. Craik, J. Phys. Chem. B, 2012, 116, 6097–6105 CrossRef CAS PubMed.
- K. Wittayanarakul, S. Hannongbua and M. Feig, J. Comput. Chem., 2008, 29, 673–685 CrossRef CAS PubMed.
- G. Holzwarth and P. Doty, J. Am. Chem. Soc., 1965, 87, 218–228 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- P. Han, K. Wang, X. Dai, Y. Cao, S. Liu, H. Jiang, C. Fan, W. Wu and J. Chen, Mar. Drugs, 2016, 14, 213 CrossRef PubMed.
- B. R. Green, M.-M. Zhang, S. Chhabra, S. D. Robinson, M. J. Wilson, A. Redding, B. M. Olivera, D. Yoshikami, G. Bulaj and R. S. Norton, FEBS J., 2014, 281, 2885–2898 CrossRef CAS PubMed.
- J. R. McArthur, G. Singh, M. L. O'Mara, D. McMaster, V. Ostroumov, D. P. Tieleman and R. J. French, Mol. Pharmacol., 2011, 80, 219–227 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06103c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Yanyan Chub,
Shengbiao Wanab,
Tao Jiangab and
Rilei Yu
ab,
Yanyan Chub,
Shengbiao Wanab,
Tao Jiangab and
Rilei Yu