
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of silacyclopent-2-en-4-ols via intramolecular [2 + 2] photocycloaddition of benzoyl(allyl)silanes†
João R.
Vale
ab,
Arto
Valkonen
 c,
Carlos A. M.
Afonso
*b and
Nuno R.
Candeias
*a
c,
Carlos A. M.
Afonso
*b and
Nuno R.
Candeias
*a
aFaculty of Engineering and Natural sciences, Tampere University, Korkeakoulunkatu 8, 33101 Tampere, Finland. E-mail: nuno.rafaelcandeias@tuni.fi
bInstituto de Investigação do Medicamento (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal. E-mail: carlosafonso@ff.ulisboa.pt
cDepartment of Chemistry, University of Jyvaskyla, P.O. Box 35, 40014 Jyväskylä, Finland
First published on 12th October 2019
Abstract
Organosilicon compounds are versatile units with a wide range of uses from medicinal chemistry to the field of organic electronics. An unprecedented method for the synthesis of novel diaryl-substituted silacyclopentenols via blue-light promoted intramolecular [2 + 2] photocycloaddition of acyl silanes is herein disclosed. Additionally, the present findings demonstrate the influence of the olefin substituents in controlling the regioselectivity of the intramolecular Paternò–Büchi reaction, providing silacycles different from previously reported ones. The high degree of functionalization of these compounds makes them attractive precursors to other synthetically challenging silacyclopentanes.
Introduction
The incorporation of silicon into an organic molecule can alter some of its physical and chemical properties, making organosilicon compounds attractive candidates for medicinal chemistry.1,2 Although silicon is an isostere of sp3-hybridised carbon, replacing carbon by silicon can: alter reactivity, increase lipophilicity, induce different conformations and increase ring sizes.1 Organosilicons have the tendency to form penta- and hexa-coordinated species3,4 and labile Si–O, Si–N and Si–H bonds in aqueous media. Moreover, polarised C–Si bounds have increased reactivity as compared to C–C bounds. Because of these features, organosilicon compounds have shown considerable potential in the medicinal chemistry field over the past 55 years.1 The virtually simple replacement of carbon by silicon in a known drug or scaffold has been widely explored in the design of new bioactive compounds.5,6 Drastic changes in the pharmacological profiles of silicon bioisoster of venlafaxine,7 haloperidol8 and bexarotene9 have been previously reported, including improvements in the selectivity profile or lower toxicity of the drug metabolites.Notwithstanding the role of silicon in drug discovery,2 organosilicon compounds have also gained notoriety in molecular electronics10 and polymer science.11 Conjugated organosilicon materials are particularly promising for the organic electronics and photonics field.12 The insertion of silicon atoms in conjugated organic motifs changes the HOMO and LUMO energy levels, creating a new broad class of important semiconductors. The 5-membered conjugated silacycle silole is a particularly interesting building block in this research area and in the development of silicon-containing polymers.13 Given the natural abundance of silicon and its lack of intrinsic toxicity, processes targeting the synthesis of organosilanes14 and the modification of C–Si bonds are becoming more available.15 Despite the versatility and importance of silacycles, methods for their preparation are somewhat scarce.16 Silacyclobutanes (SCBs) are generally synthetised by the convenient intramolecular silylation of in situ prepared Grignard reagents.16 Due to their high ring strain and Lewis acidity, SCBs are often used as precursors to other organosilicon compounds through ring opening/expansion reactions catalysed by transition metals.16,17 Even though ring expansion of SCBs can also provide silacyclopentanes under specific conditions, the most direct method for their synthesis consists in the addition of metal treated 1,3-butadienes to dichlorosilanes.18,19 The synthesis of silacyclopentenes through palladium-catalysed silylene-1,3-diene [4 + 1] cycloaddition20 allows flexibility on substitution of the cyclopentene core.
Considering the importance of the cyclopentane motif in bioactive compounds,21–26 we reasoned that new methods for the preparation of 5 membered-ring silacycles are needed for several fields of molecular sciences. From a synthetic perspective, achieving the cyclopentene moiety would be preferable due to potentially further functionalization of the double bound, such as asymmetric oxidative transformations.27,28 Silacyclopent-2-enes are especially challenging as the ring is usually constructed from a highly functionalised silane (Scheme 1a),29–31 imposing limitations on the degree of substitution on the cyclic core. Further modifications of silacyclopent-2-enes have also been reported32–35 but additional functionalization of the cyclopentene ring is rare and synthetic flexibility on olefin substitution is often limited. Amongst the aforementioned scaffold, silacyclopent-2-enes bearing an allylic alcohol are of particular interest. Tomooka's group has utilised such compounds in the synthesis of various functionalised silacylclopentanes, some presenting biological activity, via Mitsunobu, Tsuji–Trost, and other transformations.34,35 Additionally, similar silacyclopent-2-en-4-ols have been used in the synthesis of a polyol motif present in several natural products.36
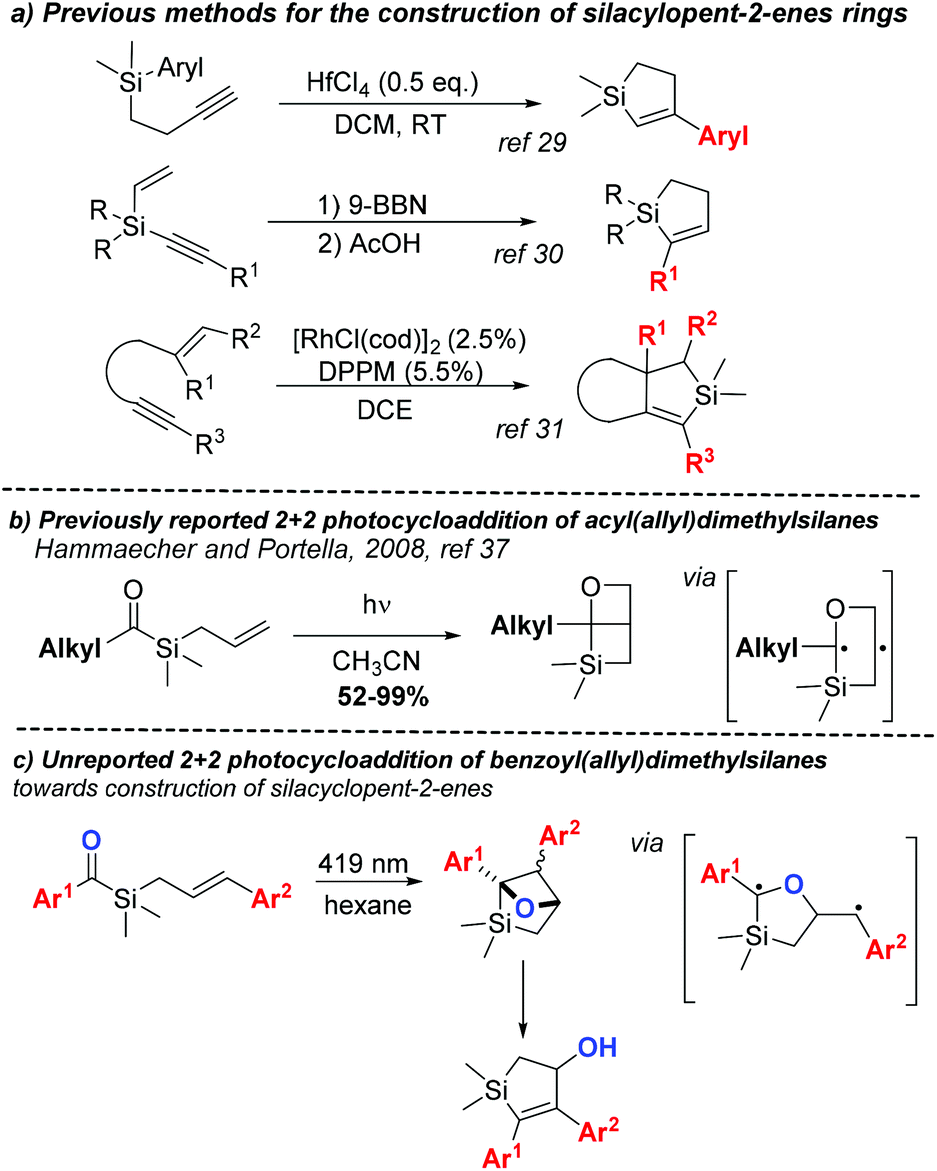 | ||
| Scheme 1 Methods for construction of silacyclopent-2-enes and photocycloaddition of acylsilanes. | ||
In 2008, Portella et al. reported a photochemical [2 + 2] cycloaddition of acylsilanes (Scheme 1b).37 Upon irradiation, acyl(allyl)silanes undergo intramolecular Paternò–Büchi reaction, producing bicyclic oxetanes in moderate to good yields and providing a new method for the synthesis of SCBs. While new 1-alky-6-oxa-2-silabicyclo[2.2.0]hexanes could be prepared by this way, irradiation of benzoyl(allyl)silane resulted in an intractable reaction mixture. Since the relative stability of the triplet biradical intermediates in the Paternò–Büchi reaction plays an important role in its stereo- and regioselectivity,38,39 we envisioned that substitution on the olefin moiety with a radical stabilising group could tame the reactivity of the intermediates, altering the reaction profile observed by Portella. This could alter the regioselectivity of the cycloaddition, and the subsequent oxetane ring-opening would eventually yield silacyclopentenols (Scheme 1c).
Acylsilanes have attracted immense scientific interest since their first preparation by Brook in 1957.40 Despite the extensive research conducted on these compounds in the past few decades,41–43 novel methodologies that utilize acylsilanes as powerful reagents in organic chemistry are still being reported.44–46 Experimentally, the use of aromatic acylsilanes is particularly attractive as they do not undergo side reactions involving the carbonyl α-carbon.47 Moreover, they show an absorption maximum at around 425 nm, thus allowing photoexcitation using visible blue light. The handling of acylsilanes requires special precautions as they are known to undergo Brook rearrangement to a reactive carbene after photo-irradiation or thermal conditions. This carbene has been shown to undergo insertion reactions in polarised heteroatom-hydrogen bounds,48,49 cycloadditions to aldehydes50 and alkynes,51,52 cross-coupling reaction with organoboronic esters53 and even C–H insertions54–56 under harsher conditions. Carbene formation and its subsequent reactions are then possible competitive destructive pathways to the [2 + 2] photocycloaddition. This influence can be minimised by using dry, aprotic solvents and by protecting free alcohols or amines in the acylsilane starting material.
Results and discussion
To investigate our hypothesis, cinnamyl silane 3a was synthesised (Scheme 2). Initial investigations using the dithiane umpolung approach failed as the harsh conditions required for the deprotection of the dithiane moiety were incompatible with the olefin group. Inspired by Portella's success in the synthesis of acyl(allyl)silanes using benzotriazole hemiaminals as umpolung equivalents of aldehydes,37,57 we adopted a similar synthetic pathway. This strategy, originally developed by Katritzky,58 benefits from facile late-stage hydrolysis of the hemiaminals to the corresponding carbonyls. Olefin cross-metathesis was performed on intermediate 2a, as the acylsilane moiety seems to inhibit the Grubbs catalyst.57 Gratifyingly, 3a was obtained in reasonable 53% yield after hydrolysis. Compound 3a was irradiated with blue-light for 5 hours in dry hexane, and complete consumption of the starting material was observed (Scheme 3). NMR analysis showed that oxetane 4a was cleanly obtained as a mixture of two diastereoisomers (syn/anti = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), resulting from the [2 + 2] photocycloaddition reaction. Notably, reverse regioselectivity in the Paternò–Büchi reaction was observed by Portella et al.37 for unsubstituted acyl(allyl)silanes (Scheme 1b).
:2), resulting from the [2 + 2] photocycloaddition reaction. Notably, reverse regioselectivity in the Paternò–Büchi reaction was observed by Portella et al.37 for unsubstituted acyl(allyl)silanes (Scheme 1b).
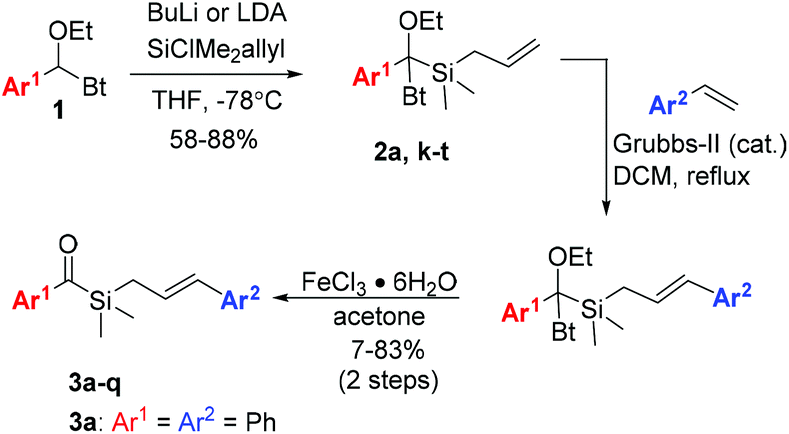 | ||
| Scheme 2 Synthesis of benzoylsilanes 3. | ||
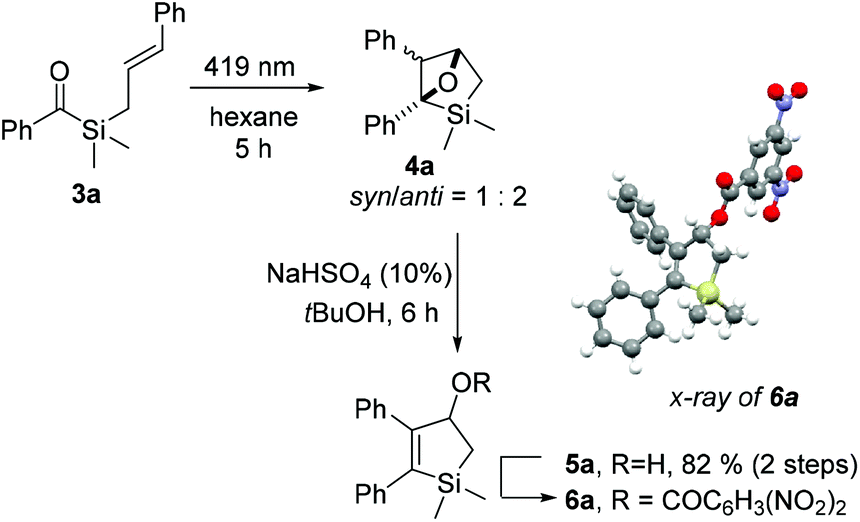 | ||
| Scheme 3 [2 + 2] photocycloaddition of cinnamyl silane 3a, acid-catalysed isomerization to 5a, and crystal structure of 3,5-dinitrobenzoylated 6a. | ||
Compound 4a proved to be unstable to silica column purification and overall acidic conditions, producing alcohol 5a and other products. After optimization, we found that treating 4a with catalytic NaHSO4 in tert-butanol leads to complete and selective isomerization to 5a, in 82% overall yield. Its structure was proven through esterification with 3,5-dinitrobenzoylchloride to yield 6a and following single crystal X-ray diffraction analysis of the obtained ester.
Realising the potential of the photocycloaddition–isomerization sequence as a direct and selective way to obtain 2,3-arylsubstituted silacyclopent-2-en-4-ols, we synthesised a series of acylsilanes 3 to be submitted to the same transformation (Scheme 2). Sila-hemiaminals 2 were obtained in very good yields, containing a wide range of functional groups in the aryl moiety. Nitro-substituted aromatics however failed to add to the silylchloride reagent, presumably due to the low nucleophilicity of the lithiated intermediate. Issues related with the instability of the lithiated anion of 1 bearing electron-rich groups in Ar1 were overcome by tBuLi addition to a stirring solution of 1 and silyl chloride. The syntheses proceeded with cross-metathesis of intermediates 2 with styrenes. Low to moderate yields were obtained in some cases due to incomplete conversion of 2, as the dimerization of styrene to stilbene was a fast competitive reaction. 2-Pyrididyl, 4-cyano- and 4-dimethylamine-benzene resulted in complete deactivation of the catalyst, as no reactions were observed after several days at reflux. The detrimental effect of nitrogen bases and N-heterocycles in metathesis reaction is known and still a problematic issue in this field.59
Despite the synthetic challenges, a series of seventeen aryl-substituted acylsilanes 3 were obtained and tested in the intramolecular [2 + 2] photocycloaddition reaction (Scheme 4). The reaction scope appears to be quite vast, withstanding electron withdrawing and electron donating substituents, as well as ortho, meta and para substitution. Gratifyingly, both variations at 3- and 2-positions of the 2,3-diaryl-silacyclopent-2-ene-4-ol could be performed. The reaction tolerates the presence of substituents prone for further derivatization such as acetyl (5e), nitrile (5g), aldehyde (5h) and silyl ether (5n). Formation of sulfur heterocycle derivative 5o contrasts with its oxygen homologue 5p, for which only traces could be observed. Also ortho-methyl substitution on the ketone side (Ar1) led to the recovery of starting material even after 8 hours of irradiation.
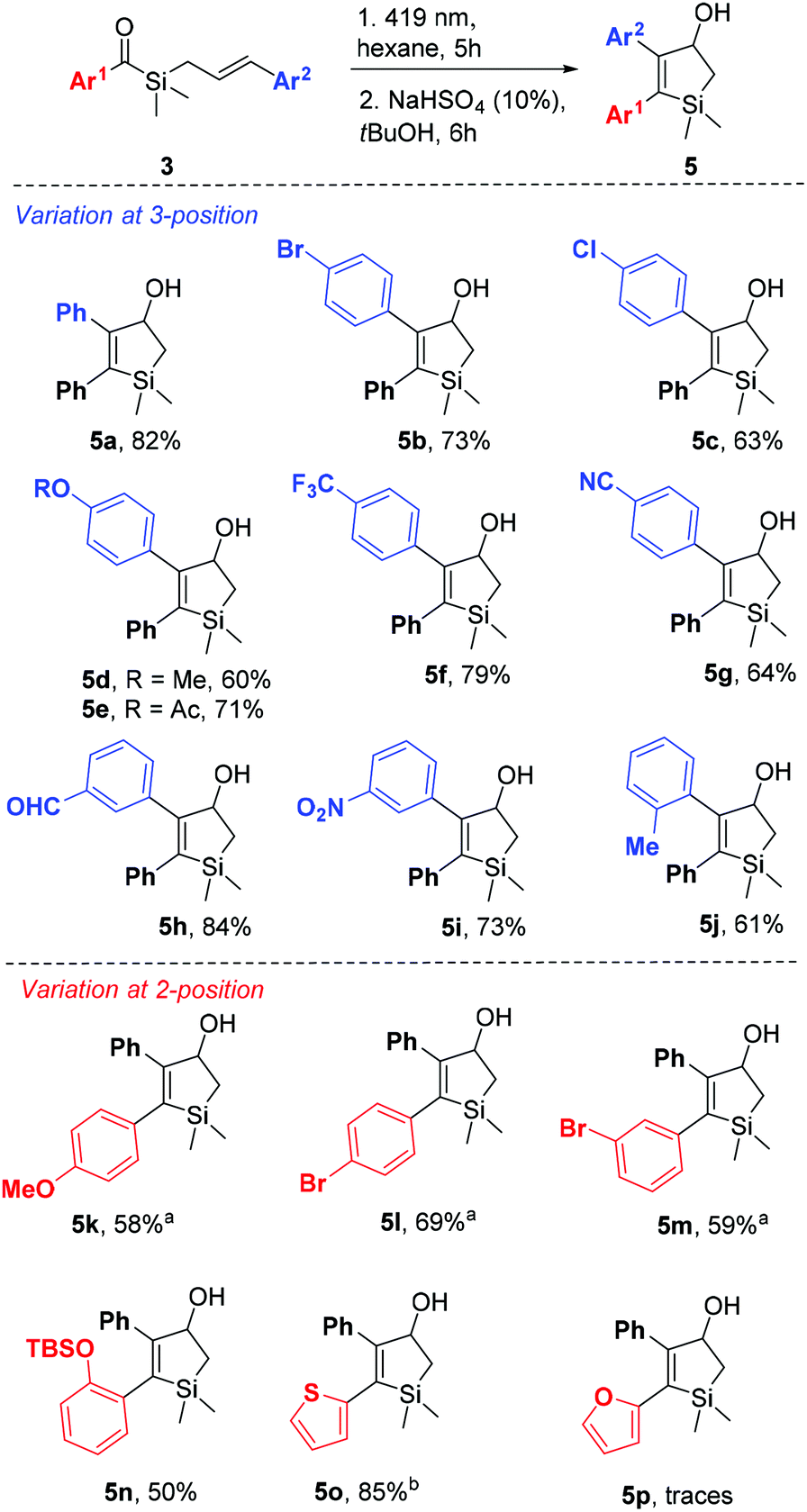 | ||
| Scheme 4 Scope of the [2 + 2] photocycloaddition of benzoylsilanes 3. aPPTS (10%) was used instead of NaHSO4. bNo acid treatment was needed. | ||
In order to better understand the dramatic change in regioselectivity of the [2 + 2] photocycloaddition of 3a compared to its terminal olefin analogues, DFT calculations were performed and the energies of the possible biradical intermediates compared (see ESI† for details). The Paternò–Büchi photocycloaddition typically occurs through reaction of a carbonyl compound in the excited state with an alkene in the ground state. Time dependent DFT studies on 3a and 7 show that the first vertical transition corresponds to populating the singlet excited state with comparable energies for both molecules (ca. 64 kcal mol−1, Scheme 5a), through a carbonyl n → π* transition between HOMO−1 and LUMO, as can be observed in the case of 3a (Scheme 5b). As triplet excited states for both molecules are generally ca. 11 kcal mol−1 more stable than their singlet counterparts, they likely undergo intersystem spin crossing. Analysis of the molecular orbitals of 3aT1 verifies that LSOMO and HSOMO correspond to the half-occupied n and π* carbonyl orbitals, respectively (Scheme 5c).
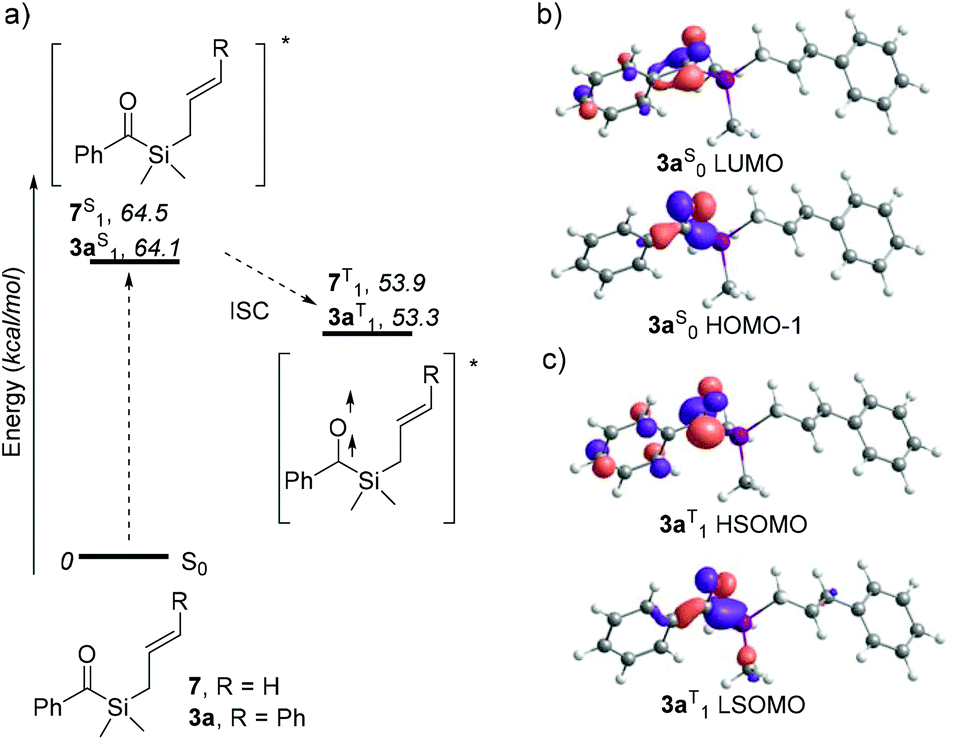 | ||
| Scheme 5 (a) Energy profile for excited states population of acylsilanes 3a and 7. Free energies of the species are indicated in italics (kcal mol−1), referring to corresponding acylsilane in ground state. (b) Representation of most significant molecular orbitals involved in first vertical excitation of 3a. (c) Single occupied orbitals of 3a. | ||
The two paths concerning the attack of the carbonyl oxygen of the triplet excited state to the olefin moiety of the allylsilane were considered‡ (Scheme 6). Comparing the formation of the two biradical species upon either exo and endo attacks of the oxygen to the cinnamyl moiety, a preference would be expected towards the former path due to stabilization of the radicals by the aromatic ring in 3a:br5. The less than 1 kcal mol−1 difference in the stabilities of the biradicals derived from 7 nevertheless suggests two competing mechanisms. Since, when starting from aliphatic acyl(allyl)silanes, Portella et al. observed the exclusive formation of the 6-endo-trig cyclization-derived product (Scheme 1b), benzoylsilane 7 was synthesised and irradiated in order to determine the effect of the carbonyl substituent on the reaction outcome.
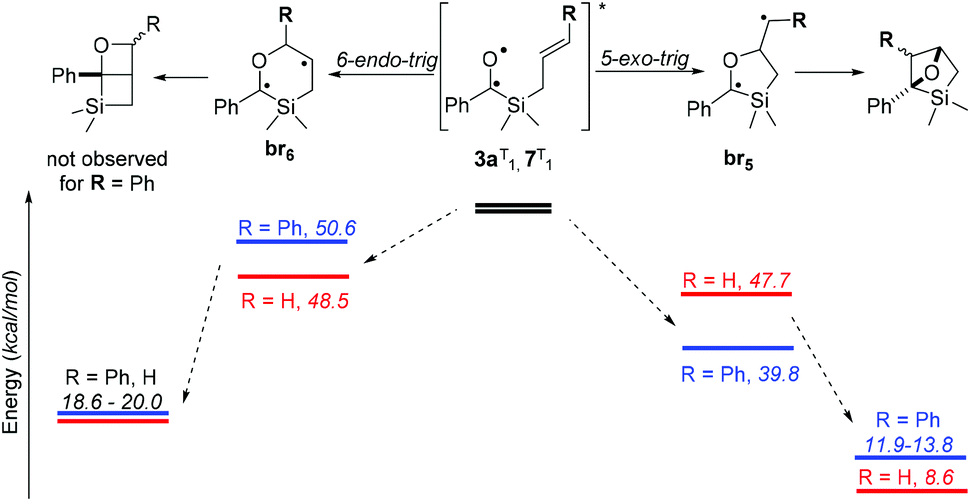 | ||
| Scheme 6 Proposed mechanism for the intramolecular [2 + 2] photocycloaddition of acylsilanes. | ||
After irradiation of 7 in hexane for 8 hours, we observed complete and clean conversion to the structural isomers 8 and 9, in 1 to 1.5 ratio, respectively (Scheme 7). Both compounds proved to be unstable to silica column chromatography. Indirect detection of compound 9 was possible after acid treatment with PPTS and isomerization to the isolable allylic alcohol 10 in 41% overall yield, which was further functionalised with 3,5-dinitrobenzoyl chloride to give ester 11 for X-ray diffraction analysis (Scheme 7). Despite the many attempts in isolating compound 8 or its decomposition products, we could only verify its presence through identification of characteristic peaks in 1H and 13C NMR spectra of the reaction mixture, in accordance with previous oxetanes characterization by Portella et al.37 (Scheme 1b).
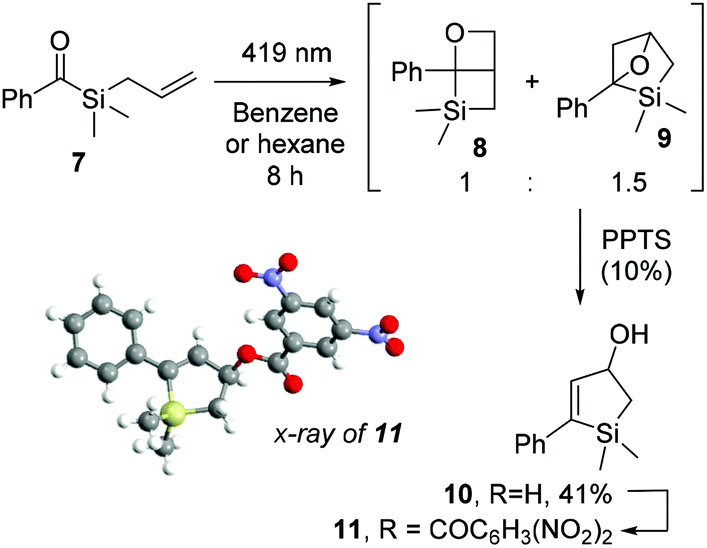 | ||
| Scheme 7 Irradiation of benzoyl(allyl)silane 7 and its non-regioselective [2 + 2] cycloaddition. | ||
Experimentally, the lack of regioselectivity for the cycloaddition of compound 7 is in agreement with its DFT predicted reactivity, highlighting the need of a radical stabilising group connected to the olefin in order to promote complete regioselectivity towards the 5-exo-trig pathway.
In order to demonstrate the versatility of these unprecedented 2,3-disubstituted silacyclopentenols as precursors to other silacyclopentanes, compound 5a was subjected to known transformations (Scheme 8). Diastereoselective epoxidation with m-CPBA quantitatively delivered 12a, a candidate precursor to a series of polyols. Ethanolysis of a transient carbocation formed after acid treatment of 5a yielded ether 13a. In addition, olefin reduction with 1 atm of hydrogen in the presence of catalytic palladium on carbon quantitatively yields the silacyclopentanol 14a as mixture of two diastereoisomers (syn/anti = 1.4:1). Finally, inspired by the structural similarity of selective COX-2 enzyme inhibitors60 derived from cyclopentane, the removal of the hydroxyl functionality of 5a was attempted. Gratefully, use of Yamamoto's HSiEt3/catalytic B(C6F5)3 system61 allowed deoxygenation to 15a in reasonable yield. The use of such compounds as bioisosteres of cyclopentanes could pave the way in the adoption of the new intramolecular [2 + 2] photocycloaddition in the silicon/carbon switch strategy. Oxidation of allylic alcohol 5a was accomplished using excess MnO2 in cold DCM, giving ketone 16a quantitatively. Low temperatures are vital as 16a proved to easily isomerize to the silyl enol ether 17a under thermal conditions.
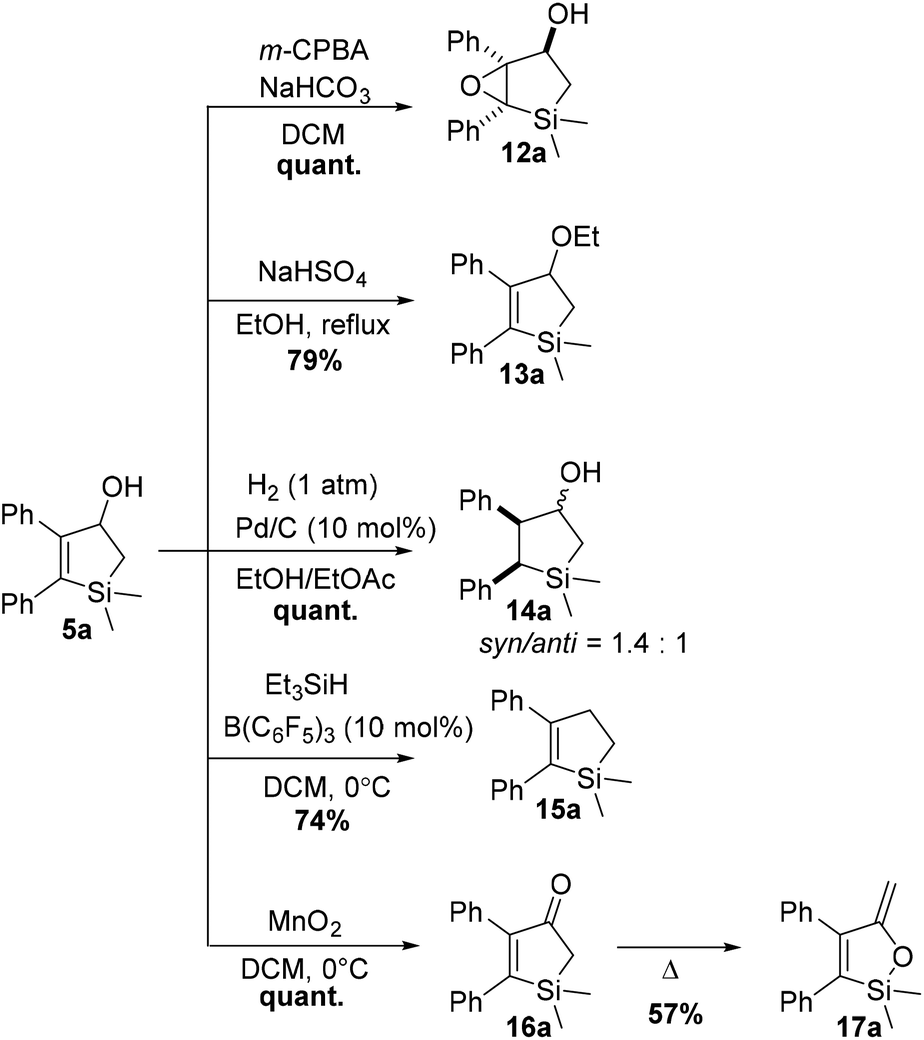 | ||
| Scheme 8 Examples of chemical transformations of 5a. | ||
Conclusions
In conclusion, we have developed a methodology for the synthesis of novel 2,3-diaryl silacyclopent-2-en-4-ols. This strategy involved the synthesis of olefin-substituted benzoyl(allyl)silanes through the benzotriazole umpolung and metathesis reactions. Blue-light irradiation of these molecules promotes a [2 + 2] photocycloaddition reaction with complete regioselectivity, governed by the stability of the biradical intermediate as supported by computational calculations. Acid-promoted isomerization yields, to the best of our knowledge, the so far unavailable 2,3-disubstituted silacyclopentenols. Moreover, this study expands the rather short collection of intramolecular Paternò–Büchi reaction examples while demonstrating the influence of the olefin substituents in controlling the regioselectivity of such reaction. The obtained compounds may be used as versatile precursors to other silacyclopentanes that could find their use in the silicon/carbon switch approach in medicinal chemistry as bioisosters of bioactive cyclopentanes, or as organosilicon motifs in organic electronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Academy of Finland is duly acknowledged for financial support to N. R. C. (Decisions No. 326487 and 326486) and to A. V. (No. 314343). Fundação para a Ciência e Tecnologia (FCT) is acknowledged for financial support to JRV (SFRH/BD/120119/2016). CSC-IT Center for Science Ltd, Finland is acknowledged for the allocation of computational resources.Notes and references
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef PubMed.
- A. D. Dilman and S. L. Ioffe, Chem. Rev., 2003, 103, 733–772 CrossRef CAS PubMed.
- S. Rendler and M. Oestreich, Synthesis, 2005, 1727–1747 CAS.
- R. Tacke and S. Dörrich, Atypical Elements in Drug Design, 1st edn, 2014 Search PubMed.
- R. Ramesh and D. S. Reddy, J. Med. Chem., 2017, 61, 3779–3798 CrossRef PubMed.
- O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, J. B. H. Warneck and R. Tacke, Organometallics, 2006, 25, 1188–1198 CrossRef.
- R. Tacke, F. Popp, B. Müller, B. Theis, C. Burschka, A. Hamacher, M. U. Kassack, D. Schepmann, B. Wünsch, U. Jurva and E. Wellner, ChemMedChem, 2008, 3, 152–164 CrossRef CAS PubMed.
- J. B. Bauer, W. P. Lippert, S. Dörrich, D. Tebbe, C. Burschka, V. B. Christie, D. M. Tams, A. P. Henderson, B. A. Murray, T. B. Marder, S. A. Przyborski and R. Tacke, ChemMedChem, 2011, 6, 1509–1517 CrossRef CAS PubMed.
- T. A. Su, H. Li, R. S. Klausen, N. T. Kim, M. Neupane, J. L. Leighton, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Acc. Chem. Res., 2017, 50, 1088–1095 CrossRef CAS PubMed.
- R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC.
- S. A. Ponomarenko and S. Kirchmeyer, Adv. Polym. Sci., 2011, 235, 33–110 CrossRef.
- J. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714–1742 CrossRef CAS.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- Q. C. Mu, J. Chen, C. G. Xia and L. W. Xu, Coord. Chem. Rev., 2018, 374, 93–113 CrossRef CAS.
- H. Chen, Y. Chen, X. Tang, S. Liu, R. Wang, T. Hu, L. Gao and Z. Song, Angew. Chem., Int. Ed., 2019, 58, 4695–4699 CrossRef CAS PubMed.
- G. Manuel, P. Mazerolles and E. T. J. Florence, J. Organomet. Chem., 1971, 30, 5–19 CrossRef CAS.
- G. K. Henry, R. Shinimoto, Q. Zhou and W. P. Weber, J. Organomet. Chem., 1988, 350, 3–8 CrossRef CAS.
- T. Ohmura, K. Masuda, I. Takase and M. Suginome, J. Am. Chem. Soc., 2009, 131, 16624–16625 CrossRef CAS PubMed.
- D. S. Straus and C. K. Glass, Med. Res. Rev., 2001, 21, 185–210 CrossRef CAS PubMed.
- S. M. Roberts, M. Gabriella and E. S. Sickle, J. Chem. Soc., Perkin Trans. 1, 2002, 1735–1742 RSC.
- H. Almahli, Curr. Top. Med. Chem., 2017, 17, 2903–2912 CrossRef CAS PubMed.
- Z. Sevcikova, M. Pour, D. Novak, J. Ulrichova and J. Vacek, Mini-Rev. Med. Chem., 2014, 14, 322–331 CrossRef CAS PubMed.
- B. Heasly, Curr. Org. Chem., 2014, 18, 641–686 CrossRef.
- S. P. Simeonov, P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744–5893 CrossRef CAS PubMed.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- A. Pfenninger, Synthesis, 1986, 89–116 CrossRef CAS.
- N. Asao, T. Shimada, T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 10899–10902 CrossRef CAS PubMed.
- B. Wrackmeyer, O. L. Tok and R. Kempe, Inorg. Chim. Acta, 2005, 358, 4183–4190 CrossRef CAS.
- T. Ohmura, I. Sasaki and M. Suginome, Org. Lett., 2019, 21, 1649–1653 CrossRef CAS PubMed.
- A. C. Stevens and B. L. Pagenkopt, Org. Lett., 2010, 12, 3658–3661 CrossRef CAS PubMed.
- D. Liu and S. A. Kozmin, Angew. Chem., Int. Ed., 2001, 40, 4757–4759 CrossRef CAS PubMed.
- K. Igawa, D. Yoshihiro, Y. Abe and K. Tomooka, Angew. Chem., Int. Ed., 2016, 55, 5814–5818 CrossRef CAS PubMed.
- K. Igawa, A. Kuroo, D. Yoshihiro, Y. Yamanaka and K. Tomooka, Synlett, 2017, 28, 2445–2448 CrossRef CAS.
- D. Liu and S. A. Kozmin, Org. Lett., 2002, 4, 3005–3007 CrossRef CAS PubMed.
- C. Hammaecher and C. Portella, Chem. Commun., 2008, 5833 RSC.
- M. Abe, T. Kawakami, S. Ohata and K. Nozaki, J. Am. Chem. Soc., 2004, 126, 2838–2846 CrossRef CAS PubMed.
- F. Kong, B. Zhai and Q. Song, Photochem. Photobiol. Sci., 2008, 7, 1332–1336 RSC.
- A. G. Brook, J. Am. Chem. Soc., 1957, 79, 4373–4375 CrossRef CAS.
- A. Ricci and A. Degl'Innocenti, Synthesis, 1989, 647–660 CrossRef CAS.
- P. C. B. Page, S. S. Klair and S. Rosenthal, Chem. Soc. Rev., 1990, 19, 147 RSC.
- H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540–8571 RSC.
- J. Y. Lv, Z. Xu, Z. J. Zheng, L. Li, Y. M. Cui, J. Cao, K. F. Yang and L. W. Xu, RSC Adv., 2017, 7, 54934–54938 RSC.
- F. G. Zhang and I. Marek, J. Am. Chem. Soc., 2017, 139, 8364–8370 CrossRef CAS PubMed.
- L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, ACS Catal., 2018, 8, 304–309 CrossRef CAS.
- A. G. Brook and J. M. Duff, Can. J. Chem., 1973, 51, 352–360 CrossRef CAS.
- J. M. Duff and A. G. Brook, Can. J. Chem., 1973, 51, 2869–2883 CrossRef CAS.
- H. Watanabe, T. Kogure and Y. Nagai, J. Org. Chem., 1972, 43, 285–291 CrossRef CAS.
- A. G. Brook and J. B. Pierce, Can. J. Chem., 1971, 49, 1622–1628 CrossRef CAS.
- H. J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F. F. Pan and C. Bolm, Adv. Synth. Catal., 2012, 354, 2157–2161 CrossRef CAS.
- P. Becker, D. L. Priebbenow, H. J. Zhang, R. Pirwerdjan and C. Bolm, J. Org. Chem., 2014, 79, 814–817 CrossRef CAS.
- K. Ito, H. Tamashima, N. Iwasawa and H. Kusama, J. Am. Chem. Soc., 2011, 133, 3716–3719 CrossRef CAS.
- A. R. Bassindale, A. G. Brook and J. Harris, J. Organomet. Chem., 1975, 90, C6–C8 CrossRef CAS.
- B. Fund and F. Summer, J. Org. Chem., 1982, 47, 2668–2670 CrossRef.
- Z. Shen and V. M. Dong, Angew. Chem., Int. Ed., 2009, 48, 784–786 CrossRef CAS PubMed.
- C. Hammaecher, J. Bouillon and C. Portella, Tetrahedron, 2009, 65, 5527–5534 CrossRef CAS.
- A. R. Katritzky, H. Lang, Z. Wang, Z. Zhang and H. Song, J. Org. Chem., 1995, 60, 7619–7624 CrossRef CAS.
- K. Lafaye, C. Bosset, L. Nicolas, A. Guérinot and J. Cossy, Beilstein J. Org. Chem., 2015, 11, 2223–2241 CrossRef CAS PubMed.
- D. B. Reitz, J. J. Li, M. B. Norton, E. J. Reinhard, J. T. Collins, G. D. Anderson, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert and P. C. Isakson, J. Med. Chem., 1994, 37, 3878–3881 CrossRef CAS PubMed.
- V. Gevorgyan, J. X. Liu, M. Rubin, S. Benson and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 8919–8922 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols and NMR, computational and X-ray diffraction data. CCDC 1935510 and 1935511. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo01028a |
| ‡ The possible intermediates involved in the paths derived from the attack of the carbonyl carbon to the olefin moiety were also considered, although leading to biradicals considerably higher in energy (see ESI†). |
| This journal is © the Partner Organisations 2019 |