
Mono epoxidation of α,ω-dienes using NBS in a water-soluble cavitand†
Venkatachalam
Angamuthu
 a,
Faiz-Ur
Rahman
a,
Manuel
Petroselli
a,
Yongsheng
Li
a,
Yang
Yu
*a and
Julius
Rebek
Jr.
*ab
a,
Faiz-Ur
Rahman
a,
Manuel
Petroselli
a,
Yongsheng
Li
a,
Yang
Yu
*a and
Julius
Rebek
Jr.
*ab
aCenter for Supramolecular Chemistry & Catalysis and Department of Chemistry, Shanghai University, 99 Shang-Da Road, Shanghai 200444, P. R. China. E-mail: yangyu2017@shu.edu.cn
bThe Skaggs Institute for Chemical Biology and Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: jrebek@scripps.edu
First published on 29th July 2019
Abstract
Water-soluble host molecules offer a range of environments to their guests. Polar functions of guests are exposed to the medium while hydrophobic groups are generally buried in the containers and hidden from reagents in solution. Here, we apply these preferences to convert α,ω-dienes to epoxy alkenes using cavitands as reaction vessels. Reaction of one end of a diene with NBS in water gives a bromohydrin that binds in the cavitand with the hydroxyl exposed and the remaining alkene buried. Treatment with base converts the bromohydrin to an epoxide. The reaction sequence provided up to 84% yields of monoepoxides from symmetrical dienes separated by 4 to 10 methylene groups. With 1,4-diisopropenyl benzene, a nearly quantitative yield of the monoepoxide was obtained. The application should be general for converting symmetrical hydrophobic guests to unsymmetrical, amphiphilic ones.
Container molecules are widely applied in studies of molecular recognition and reactivity in confined spaces. Their use as reaction vessels1–11 and sensors12–15 is well-developed, but as blocking groups, less so: Gibb16 introduced the concept for intermolecular competition between encapsulated isomeric esters, and Fujita17 recently applied it to intramolecular competition between olefin sites in epoxidation. The key feature of water-soluble containers18 is an open end where hydrophilic groups are exposed to the aqueous medium; the hydrophobic interiors of the cavities house nonpolar functions. In this study we use cavitand containers19,20 that have been modified for water solubility21,22 (Fig. 1). They are readily synthesized by chemical methods and used as hosts in a variety of applications.23 α,ω-Difunctional compounds with long hydrophobic chains assume folded conformations in such cavitands: if the functions are hydrophilic, they remain exposed to the aqueous medium; if the functions are hydrophobic, they move to compete for the cavitand's interior. We describe here the reactions of α,ω-dienes sequestered by cavitand 1 in aqueous (D2O) solution. Hydrophobic forces drive the dienes into the cavitand and the guest moves rapidly between conformations that best fill the space.24 Reaction at one end of the diene desymmetrizes the guest's polarity and fixes its position in the cavitand's space.
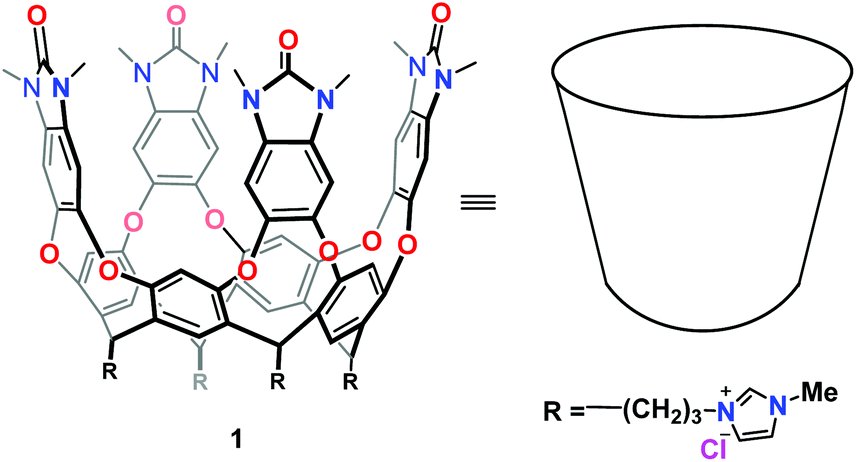 | ||
| Fig. 1 Chemical structure and schematic cartoon of the water-soluble cavitand. | ||
The partial 1H NMR spectra of α,ω-long chain dienes (C8 to C14) and 1,4-diisopropenyl benzene are shown in Fig. 2. Brief sonication of these α,ω-dienes (1.4 mM) with 1 (1.4 mM) in water (D2O) gave stoichiometric host–guest complexes. The 1H NMR signals of the guest showed characteristic upfield-shifts caused by the shielding of the aromatic panels of the host. The typical upfield shifts (−Δδ) experienced by nuclei in 1 are given in ESI (see Fig. S1†).
 | ||
| Fig. 2 (a) Partial 1H NMR (600 MHz, D2O, 298 K) spectra of the complexes formed between 1 and 2a–g. (b) Partial spectrum of 1,4-diisopropenyl benzene (3) binding with 1 and a cartoon of the host–guest complex. | ||
The conformation of linear guests inside the cavitand 1 is not fixed but moves on rapidly on the NMR chemical shift timescale. The motion may be “yo-yo” like between two J-shaped conformations or the rapid tumbling of a coiled conformation. The former is more likely for longer guests and the latter for shorter ones. In any case, the two ends of the dienes rapidly exchange positions from near the top of the cavitand to near its bottom (see Fig. S14†). The binding pattern of longer chains complies with our previous reports.25 The aromatic 1,4-disubsituted diene 3 (Fig. 2b) showed a position in which one –C(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 group is fixed deep inside the cavity. The Δδ for one CH3 is observed −4.47 ppm, near the maximum value (see Fig. S15 and S16†). The guest hydrogen signals were observed to be broad, perhaps due to some restricted motion.
CH2 group is fixed deep inside the cavity. The Δδ for one CH3 is observed −4.47 ppm, near the maximum value (see Fig. S15 and S16†). The guest hydrogen signals were observed to be broad, perhaps due to some restricted motion.
We used 1 as a chaperone to synthesize monoepoxides of dienes 2c–2f (C10–C13) and 3via electrophilic addition of NBS. As shown in Scheme 1, epoxides form via bromohydrin intermediates INT-1 and INT-2.26–29 Host–guest complexes were formed by sonication of an NMR tube containing diene and cavitand (1.4 mm) for two hours followed by addition of NBS (1 eq.). As shown in Fig. 3, the signals of 2e disappeared while the product intermediate bromohydrin increased with time. After complete conversion of the diene, aqueous (D2O) K2CO3 (0.5 eq.) was added. The conversion to 4e appeared after a reaction time of 6 hours. Comparison with authentic monoepoxide in 1 (top trace of Fig. 3a) indicated nearly no formation of guest by-products. The NMR signals of monoepoxide C12 in 1 ranged from 2.61 ppm to −2.57 ppm. This signal pattern is consistent with a fixed arrangement of the guest in the cavitand. The epoxide group is exposed, the –CH2–HCCH2 end is buried and the allyl hydrogens are deepest in the cavity.
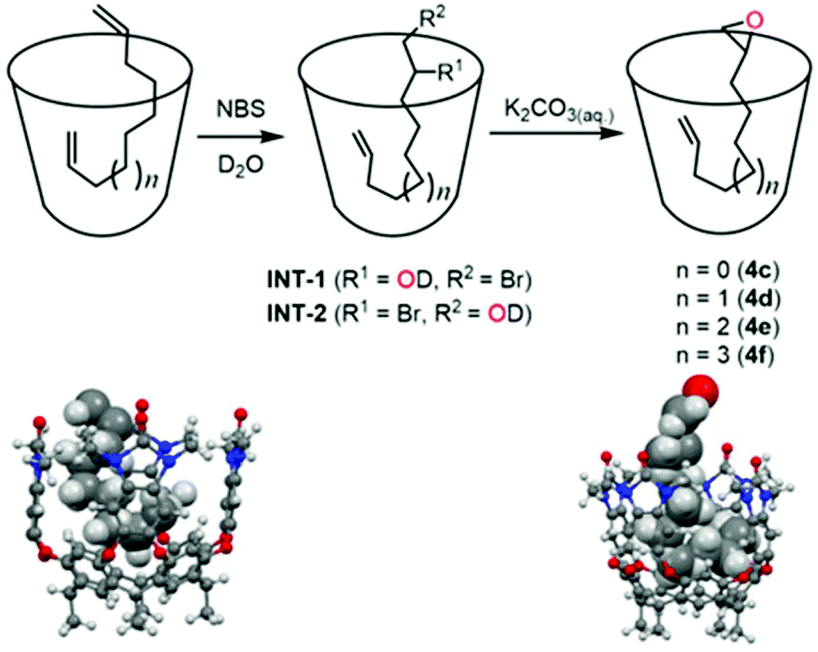 | ||
| Scheme 1 Top: Cartoons of the epoxidation process with NBS and base with cavitand 1 in aqueous medium. Bottom: Modeled complexes of α,ω-diene (C10) (left) and its monoepoxide in a J-shaped conformation (right). | ||
 | ||
| Fig. 3 Top (a): Partial 1H NMR (600 MHz, D2O, 298 K) spectra of α,ω-diene (C12) 2e in 1 recorded after addition of NBS (50 mM, 14 μL). (a) After 3 h of sonication at 25 °C with DMSO-d6 used as co-solvent; (b) 14 μL of NBS, 12 h, 50 °C; (c) sample b, 7 μL (50 mM) of K2CO3(aq), 12 h, 50 °C; (d) authentic 2-(dec-9-en-1-yl)oxirane (4e) in cavitand 1. Bottom (b): Cartoon of the conversion of (C12) to mono epoxide 4e with assignments of the product methylene signals. | ||
Parallel results were obtained with the other linear aliphatic dienes (Fig. S18–S24†). All authentic monoepoxides were synthesized by using m-CPBA (0.5 eq.) in DCM (see ESI†). This provides monoepoxides in organic solvents but the yields and selectivity are lower. The product conformations were confirmed by 2D COSY studies (see Fig. S25–S28†). The formation of the bromohydrin intermediate was unambiguously confirmed by comparison with an authentic standard in the cavitand (see Fig. S29†). The fixed conformation of the complex having the –CHCH2 end buried prevents further electrophilic reactions with the aqueous NBS.
Addition of another 0.2 equivalents of NBS to the reaction mixture after 6 h did not result in changes of the integral peaks in the spectra. Only compounds with longer lipophilic chains such as compounds 4e and 4f showed small amounts of impurities (10%) in their reactions with excess NBS (see Fig. S30†). This results confirmed that the terminal –CHCH2 group of the buried end is protected by the cavitand and inaccessible to reagents. The product yields were calculated by 1H NMR using dimethyl sulfone (DMS) as an internal standard. The yields observed were 84, 70, 64, and 57% for 2c, 2d, 2e and 2f, respectively (see Fig. S32–S35†).
The reaction of an aromatic 1,4-substituted diene 3 in 1 also gave the respective mono oxidized product as shown in Scheme 2. Again, one of the double bonds binds and is protected by cavitand (Fig. 4); the exposed double bond reacts with NBS and provides monoepoxide 5 in D2O. Without cavitand 1, this monoepoxide cannot be obtained selectively in organic solvents. The usual products are mono- and di-aldehydes due to acid catalysed rearrangements.
 | ||
| Scheme 2 Monofunctionalization of a 1,4-disubstituted aromatic diene 3 in cavitand 1. | ||
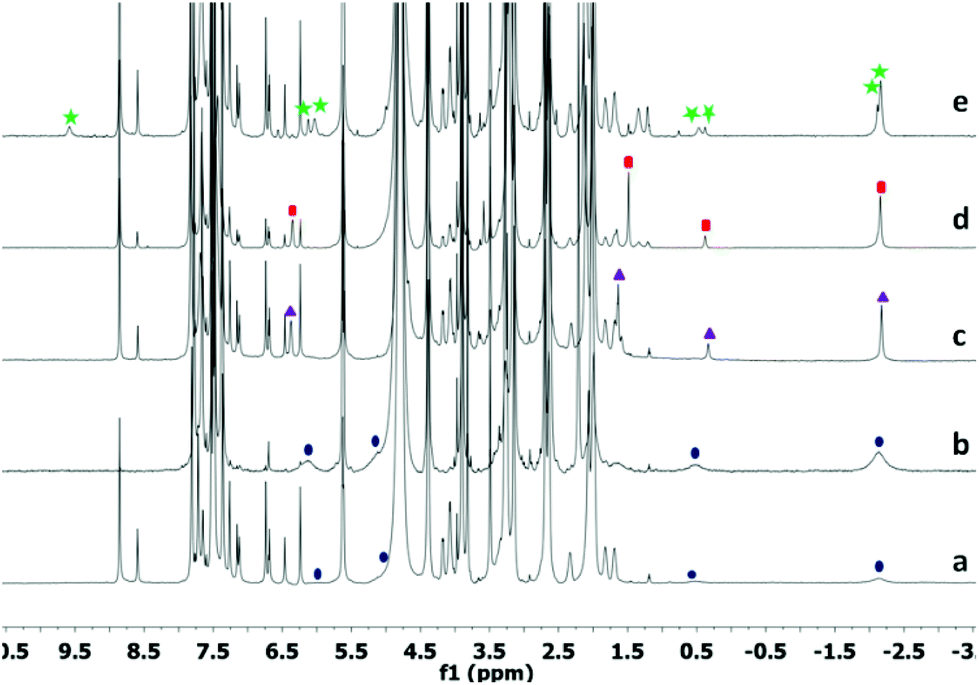 | ||
Fig. 4 Full 1H NMR spectra of 3 in 1. (a) 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host/guest complex of 3 in 1; (b) 1:1 host/guest complex; (c) reaction of sample a with NBS, stirred 2 h and stirred with K2CO3(aq.) at 50 °C for 2 h; (d) authentic mono epoxide 5; (e) authentic aldehyde 6. :1 host/guest complex of 3 in 1; (b) 1:1 host/guest complex; (c) reaction of sample a with NBS, stirred 2 h and stirred with K2CO3(aq.) at 50 °C for 2 h; (d) authentic mono epoxide 5; (e) authentic aldehyde 6. | ||
In the cavitand, aldehyde formation was not observed within 1 h as it was confirmed by binding the authentic aldehyde 6. Generally, aldehyde 6 in 1 gives two set of peaks related to hydrated and free aldehyde forms (see Fig. 4 top trace of 1H NMR). Only a trace of conversion to mono-aldehyde was observed from epoxide in 1 after few hours (8 h) (see Fig. S37†). The nearly quantitative NMR yield was calculated using hexamethylcyclotrisiloxane as internal standard (see Fig. S38†).
Unbiased control experiments are difficult to perform without the cavitand because most of the long chain dienes are practically insoluble in water. Therefore, without cavitand 1 the control experiments were performed with a solvent mixture of acetonitrile-d3 in D2O (25% v/v) and DMSO-d6 (3.6%). In these experiments, faster epoxidation reactions were observed and gave mixtures of products while using 1 equiv. of NBS (Fig. S39 and S40†). The ratio of mono, di-epoxide and starting diene were calculated by GC as 35, 45 and 18% respectively (see Fig. S41–S45†). Prolonged reaction times with 1 equiv. of NBS, the ratio remained the same as 1 h. Excess of NBS gave impurities and di-epoxides. This result highlights the striking ability of the cavitand to suppress the second electrophilic addition. In the case of aromatic compound, the formation monoepoxide or aldehyde was confirmed by gas chromatography by comparing with authentic mixtures obtained from the reaction with m-CPBA in dichloromethane (see Fig. S46–S49†). After 12 h of reaction without cavitand, control experiments with NBS (1 equiv.) gave starting diene 3, mono aldehyde 6 and di-aldehyde with the ratio of 48, 4, 38% respectively (see Fig. S50†).
In conventional solutions, the monofunctionalizations of symmetrical compounds give statistical mixtures of products if the functional sites are remote and act independently. In water-soluble cavitands as reaction vessels, we have reported a few mono functionalizations such as hydrolysis of long-chain diesters,30 the synthesis of macrocyclic ureas,24 the Staudinger reactions of diazides31 and monohydrolysis of long chain α,ω-dibromides.32 Many other organic reactions do not proceed well in aqueous medium due to the insolubility of reagents or catalysts.33 In such cases, the cavitand helps the dissolution of insoluble guests by complex formation. Cavitands may even act as enzyme mimics that can bind guests in conformations that channel reactions along pathways that fit the shape of the cavity.34–37 The present application uses differences in polarity to achieve the product selectivity and while the cavitand is used stoichiometrically, the desired products can be isolated by mere extraction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21801164), the US National Science Foundation (CHE 1801153) and by Shanghai University (N.13-G210-19-230), Shanghai, China. Dr Yang Yu thanks the Program for Professor of Special Appointment (Dongfang Scholarship) of the Shanghai Education Committee.Notes and references
- C. M. Hong, D. M. Kaphan, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 8013–8021 CrossRef CAS PubMed.
- Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS PubMed.
- D. M. Kaphan, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 9202–9205 CrossRef CAS PubMed.
- C. J. Brown, G. M. Miller, M. W. Johnson, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 11964–11966 CrossRef CAS PubMed.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2004, 43, 6748–6751 CrossRef CAS PubMed.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2004, 126, 14366–14367 CrossRef CAS PubMed.
- A. K. Sundaresan and V. Ramamurthy, Photochem. Photobiol. Sci., 2008, 7, 1555–1564 RSC.
- L. Catti, Q. Zhang and K. Tiefenbacher, Chem. – Eur. J., 2016, 22, 9060–9066 CrossRef CAS PubMed.
- J. Rebek, Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef CAS PubMed.
- D. Ajami and J. Rebek, Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS PubMed.
- A. Scarso, L. Pellizzaro, O. De Lucchi, A. Linden and F. Fabris, Angew. Chem., Int. Ed., 2007, 46, 4972–4975 CrossRef CAS PubMed.
- A. D. Gill, L. Perez, I. N. Q. Salinas, S. R. Byers, Y. Liu, B. L. Hickey, W. Zhong and R. J. Hooley, Chem. – Eur. J., 2019, 25, 1740–1745 CrossRef CAS PubMed.
- Y. Liu, L. Perez, M. Mettry, C. J. Easley, R. J. Hooley and W. Zhong, J. Am. Chem. Soc., 2016, 138, 10746–10749 CrossRef CAS PubMed.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2006, 128, 16498–16499 CrossRef CAS PubMed.
- S. Liu, H. Gan, A. T. Hermann, S. W. Rick and B. C. Gibb, Nat. Chem., 2010, 2, 847 CrossRef CAS PubMed.
- H. Takezawa, T. Kanda, H. Nanjo and M. Fujita, J. Am. Chem. Soc., 2019, 141, 5112–5115 CrossRef CAS PubMed.
- J. Murray, K. Kim, T. Ogoshi, W. Yao and B. C. Gibb, Chem. Soc. Rev., 2017, 46, 2479–2496 RSC.
- J. R. Moran, S. Karbach and D. J. Cram, J. Am. Chem. Soc., 1982, 104, 5826–5828 CrossRef CAS.
- P. Soncini, S. Bonsignore, E. Dalcanale and F. Ugozzoli, J. Org. Chem., 1992, 57, 4608–4612 CrossRef CAS.
- S. Mosca, Y. Yu, J. V. Gavette, K.-D. Zhang and J. Rebek, J. Am. Chem. Soc., 2015, 137, 14582–14585 CrossRef CAS PubMed.
- F.-U. Rahman, H.-N. Feng and Y. Yu, Org. Chem. Front., 2019, 6, 998–1001 RSC.
- Y. Yu and J. Rebek, Acc. Chem. Res., 2018, 51, 3031–3040 CrossRef CAS PubMed.
- N.-W. Wu and J. Rebek, J. Am. Chem. Soc., 2016, 138, 7512–7515 CrossRef CAS PubMed.
- N.-W. Wu, I. D. Petsalakis, G. Theodorakopoulos, Y. Yu and J. Rebek, Jr., Angew. Chem., Int. Ed., 2018, 57, 15091–15095 CrossRef CAS PubMed.
- G. Majetich, J. Shimkus and Y. Li, Tetrahedron Lett., 2010, 51, 6830–6834 CrossRef CAS.
- D. R. Dalton, V. P. Dutta and D. G. Jones, J. Am. Chem. Soc., 1968, 90, 5498–5501 CrossRef CAS.
- D. R. Dalton, R. C. Smith and D. G. Jones, Tetrahedron, 1970, 26, 575–581 CrossRef CAS.
- D. R. Dalton, J. B. Hendrickson and D. Jones, Chem. Commun., 1966, 591–592 RSC.
- Q. Shi, M. P. Mower, D. G. Blackmond and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9199–9203 CrossRef CAS PubMed.
- D. Masseroni, S. Mosca, M. P. Mower, D. G. Blackmond and J. Rebek, Angew. Chem., Int. Ed., 2016, 55, 8290–8293 CrossRef CAS PubMed.
- V. Angamuthu, M. Petroselli, F.-U. Rahman, Y. Yu and J. Rebek, Org. Biomol. Chem., 2019, 17, 5279–5282 RSC.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- P. Jagadesan, B. Mondal, A. Parthasarathy, V. J. Rao and V. Ramamurthy, Org. Lett., 2013, 15, 1326–1329 CrossRef CAS PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- A. K. Sundaresan and V. Ramamurthy, Org. Lett., 2007, 9, 3575–3578 CrossRef CAS PubMed.
- Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo00849g |
| This journal is © the Partner Organisations 2019 |