DOI:
10.1039/C9QO00831D
(Research Article)
Org. Chem. Front., 2019,
6, 3150-3157
Platinum(II)-catalyzed dehydrative C3-benzylation of electron-deficient indoles with benzyl alcohols†
Received
1st July 2019
, Accepted 12th July 2019
First published on 17th July 2019
Abstract
A synthetic strategy for the water-promoted direct dehydrative coupling of indoles with benzyl alcohols catalyzed by PtCl2(PhCN)2 in 1,2-dichloroethane has been developed. Water molecules significantly accelerate C–C bond formation, whereas the reaction proceeds slowly under anhydrous conditions. Comparative rate plots for reactions in the presence of H2O and D2O show a kinetic solvent isotope effect (KSIE) of 1.5. A Hammett study of the dehydrative reaction of para-substituted benzhydrols shows negative ρ values, indicating a build-up of cationic charge during the rate-determining sp3 C–O bond cleavage step. Our catalytic system can be used on the gram scale with simplified product isolation and catalysis is performed using 1 mol% of a Pt(II) catalyst with an additional molecule of water.
Introduction
Compared to conventional organic solvents, water possesses very interesting properties and selectivities for chemical transformations in a wide variety of carbon–carbon bond forming reactions such as the Diels–Alder reaction, aldol reaction, Claisen rearrangement and cross-coupling reactions.1,2 In 1991, the pioneering work of Breslow demonstrated the acceleration of Diels–Alder reactions by the hydrophobic effect in aqueous media.3 In 2005, Sharpless et al. demonstrated the rate acceleration of several “on water” reactions of water-insoluble substrates.4 The organic–water interface effect of dangling OH groups has been proven to enhance reaction rates. Therefore, hydrogen bonding activation and hydrophobic effects by water molecules are believed to contribute to the rate acceleration for a wide variety of organic reactions.
Indoles are valuable building blocks for pharmaceutical agents in modern drug discovery (Fig. 1).5 Therefore, efficient methods for the direct introduction of diverse functionalities on indole rings are gaining increasing interest. In recent years, direct dehydrative coupling reactions of indoles with relatively low toxic, inexpensive and abundantly available alcohols as coupling partners have emerged for the construction of C–C bonds due to the advantages of being a salt-free, environmentally benign and atom-economical process with water as the by-product.6,7
 |
| | Fig. 1 Representative biologically active 3-substituted indoles. | |
In the light of our ongoing efforts to develop dehydrative C–C bond forming reactions in water,8 we herein report the first examples of a Pt(II)-catalyzed direct dehydrative coupling reaction of electron-deficient indoles with benzyl alcohols in 1,2-dichloroethane (Scheme 1).9 In 2008, Yadav et al. reported the dehydrative benzylation of 5-nitroindole with benzylic alcohols catalyzed by phosphomolybdic acid-supported silica gel.9c While the heterogeneous catalysts offer significant advantages (e.g. simple work-up, cleaner reaction profiles and reutilization procedures), our homogeneous platinum catalyst system could be used in simple, convenient and readily available protocols for the dehydrative benzylation.
 |
| | Scheme 1 Water-promoted dehydrative C3-benzylation. | |
Notably, the PtCl2(PhCN)2 catalyst is highly effective as a Lewis acid for the activation of alcohols while Pt(II) catalysts are generally used for the π-activation of alkenes and alkynes.10 Furthermore, water molecules present in situ serve as an essential cocatalyst for achieving high catalytic activity of the Pt(II) complex under mild reaction conditions without the need for bases or other additives in the atom-economical process.
Results and discussion
Optimization of reaction conditions
Initially, electron-deficient 5-nitroindole (1a) and benzhydrol (2a) were chosen as model compounds to optimize the dehydrative reaction conditions. When using the PtCl2(PhCN)2 catalyst (5 mol%) at 50 °C for 16 h in 1,2-dichloroethane (DCE), the desired C3-benzylated product 3a was obtained selectively in 75% isolated yield (Table 1, entry 1). Excellent yield was achieved when the reaction was performed at 60 °C (entry 2, 96%). In contrast, no reaction occurred in the absence of the catalyst (entry 3). With regard to the platinum(II) catalyst, PtCl2(PhCN)2 gave the best result (entry 1 vs. entries 4–9).11 Replacing DCE with CHCl3 as a solvent resulted in almost the same yield (entry 10, 72%). In contrast, the use of CH2Cl2 or chlorobenzene afforded lower yields (entries 11 and 12). Other organic solvents such as toluene, EtOAc or 1,4-dioxane resulted in slightly lower yields (entries 13–15). No reaction occurred when polar organic solvents such as EtOH or DMSO12 were used (entries 16 and 17). The use of only water as a solvent was also disfavored in this catalytic system (entry 18).
Table 1 Effects of catalysts and solventsa
|

|
| Entry |
Pt catalyst |
Solvent |
T (°C) |
Yield (%) |
|
Reaction conditions: 5-nitroindole 1a (1 mmol), Pt catalysts (0.05 mmol), benzhydrol 2a (1.2 mmol), and solvent (4 mL), 50–80 °C, 16 h under air. Yields of the isolated products are shown.
NMR yield.
|
| 1 |
PtCl2(PhCN)2 |
DCE |
50 |
75 |
| 2 |
PtCl2(PhCN)2 |
DCE |
60 |
96 |
| 3 |
None |
DCE |
50 |
0 |
| 4 |
PtCl2 |
DCE |
50 |
0 |
| 5 |
PtCl2(PPh3)2 |
DCE |
50 |
0 |
| 6 |
Pt(COD)Cl2 |
DCE |
50 |
0 |
| 7 |
Pt(acac)2 |
DCE |
50 |
0 |
| 8 |
Na2PtCl4·xH2O |
DCE |
50 |
Trace |
| 9 |
cis-PtCl2(NH3)2 |
DCE |
50 |
0 |
| 10 |
PtCl2(PhCN)2 |
CHCl3 |
50 |
72 |
| 11 |
PtCl2(PhCN)2 |
CH2Cl2 |
50 |
15b |
| 12 |
PtCl2(PhCN)2 |
PhCl |
50 |
39 |
| 13 |
PtCl2(PhCN)2 |
Toluene |
60 |
80 |
| 14 |
PtCl2(PhCN)2 |
EtOAc |
60 |
71 |
| 15 |
PtCl2(PhCN)2 |
1,4-Dioxane |
60 |
71 |
| 16 |
PtCl2(PhCN)2 |
EtOH |
60 |
0 |
| 17 |
PtCl2(PhCN)2 |
DMSO |
60 |
0 |
| 18 |
PtCl2(PhCN)2 |
Water |
60 |
38 |
Effect of water on the dehydrative reaction
To gain insight into the effect of water on the dehydrative coupling of 1a (1 mmol) with 2a in 1,2-dichloroethane, molecular sieves (4 Å MS, 160 mg) were added to the reaction mixture to scavenge the water in the dehydration step.13 As expected, compared to the absence of 4 Å MS, the reaction rate was significantly slower (Fig. 2).14 In contrast, the addition of water (1 mmol) led to an acceleration of the reaction rate, and the reaction proceeded to near completion within 6 h.15 The rate of the reaction was faster in the presence of H2O than that in the presence of D2O with a kinetic solvent isotope effect (KSIE) of 1.5 (Fig. 3).16 This low deuterium isotope value indicates that hydrogen bonding activation plays an important role in our catalytic system.1b,17
 |
| | Fig. 2 The effect of water on the dehydrative coupling. Reaction conditions: 5-nitroindole 1a (1 mmol), PtCl2(PhCN)2 (0.05 mmol), benzhydrol 2a (1.2 mmol), and 1,2-dichloroethane, 60 °C. | |
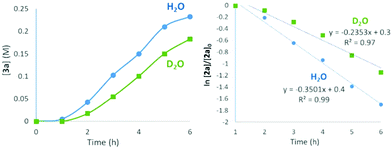 |
| | Fig. 3 Comparison of reaction rates in the presence of water and D2O. Reaction conditions: 5-nitroindole 1a (1 mmol), PtCl2(PhCN)2 (0.05 mmol), benzhydrol 2a (1.2 mmol), and 1,2-dichloroethane (4 mL) with H2O or D2O (1 mmol), 60 °C. | |
Next, the same technique was used to determine the influence of the amount of water. The observed initial rate acceleration did not depend on the amount of water added, since the excess water molecules would react with the benzyl cation intermediate. Water promoted the reaction with rates in the following order: H2O (1 mmol) > H2O (2 mmol) > D2O (1 mmol) > H2O (0.5 mmol) = no additive (0.18 mmol of water molecules would be formed after 3 h) > 4 Å MS (anhydrous conditions) > H2O (22 mmol) (Table 2, entries 1–7). Indeed, replacing 1,2-dichloroethane with only water as the solvent resulted in a lower yield (see Table 1). EtOH was found to be less effective than water (entry 2 vs. entry 8). In the absence of the Pt(II) catalyst, the addition of only water or aqueous HCl was not effective (entries 9 and 10). Other salts such as Cu(II), Ni(II) and Co(II) were less effective than Pt(II) (entries 11–13). These results indicate that the Pt(II) catalyst is highly effective for the water-promoted coupling reaction.
Table 2 Effects of the amounts of water and catalystsa
|

|
| Entry |
Catalyst |
Additive (mmol) |
NMR yield (%) |
|
Reaction conditions: 1a (1 mmol), PtCl2(PhCN)2 (0.05 mmol), 2a (1.2 mmol), and 1,2-dichloroethane (4 mL), 60 °C, 3 h under air.
DCE (3.6 mL) and H2O (0.4 mL, 10% v/v) were used as solvents.
160 mg.
HCl aq. (3 mol L−1, 18 μL) was used.
|
| 1 |
PtCl2(PhCN)2 |
H2O (0.5) |
18 |
| 2 |
PtCl2(PhCN)2 |
H2O (1) |
41 |
| 3 |
PtCl2(PhCN)2 |
H2O (2) |
26 |
| 4 |
PtCl2(PhCN)2 |
H2O (22)b |
7 |
| 5 |
PtCl2(PhCN)2 |
D2O (1) |
22 |
| 6 |
PtCl2(PhCN)2 |
None |
18 |
| 7 |
PtCl2(PhCN)2 |
4 Å MSc |
10 |
| 8 |
PtCl2(PhCN)2 |
EtOH (1) |
26 |
| 9 |
None |
H2O (1) |
0 |
| 10 |
None |
HCl aq. (1)d |
4 |
| 11 |
CuCl2 |
H2O (1) |
4 |
| 12 |
NiCl2·6H2O |
H2O (1) |
0 |
| 13 |
CoCl2·6H2O |
H2O (1) |
0 |
Based on the literature reports of the Shilov reaction,18 we propose a catalytic pathway for the water-promoted sp3 C–O bond activation of alcohol 2a as follows (Scheme 2). Initially, a ligand exchange of PtCl2(PhCN)2 with water molecules generates PtCl2(OH2)2.11,19 This aqua complex with benzhydrol (2a) is in fast equilibrium with its Lewis acid adduct A (conjugate acid). The water molecules promote the facile sp3 C–O bond cleavage, thus allowing for a much lower energy for the TS. Following bond cleavage, benzyl cation species B is formed via the rate-determining step. Shinokubo and Oshima used theoretical calculations to elucidate the importance of hydration of the hydroxy group for the smooth generation of the π-allylpalladium species for the dehydrative Tsuji–Trost reaction.20 Cozzi et al. reported that the direct generation of carbocations from alcohols is driven by the hydrogen bonding activation.21 The kinetic solvent isotope effect (KSIE) of 1.5 is consistent with the hydrogen bonding activation pathway.
 |
| | Scheme 2 Proposed pathway for the sp3 C–O bond cleavage. | |
Furthermore, Pt(II) complexes have been investigated not only for the development of synthetic methods but also as antitumor drugs. Cisplatin, cis-[PtCl2(NH3)2], is activated inside the cell through the hydrolysis of the Pt–Cl bonds to form a cationic Pt(II) aqua complex, cis-[PtCl(NH3)2(H2O)]+.22 Therefore, investigation on water-promoted reactions using our catalytic system would be important for understanding the activation process of cisplatin in the cell and rationally designing improved anticancer drugs.
Reaction scope
With the optimized conditions in hand, we examined the substrate scope of the dehydrative coupling reaction (Scheme 3). The electron-withdrawing (NO2, CO2H, CN and Cl) groups on the benzene ring of substituted indoles 1 were tolerated well to produce the corresponding products 3b–i in moderate to excellent yields (53–95%). Sterically demanding carboxy and phenyl groups of 2-substituted indoles were also tolerated in the direct substitution (65–74%, 3j–k). The scope of alcohols 2 was examined next with 4-nitroindole (1a) as the coupling partner. The use of benzhydryl alcohols with electron-donating methoxy and methyl groups, or electron-withdrawing fluoro, chloro and trifluoromethyl groups, resulted in excellent yields (72–98%, 3l–r). Furthermore, α-methylbenzyl alcohols, allylic alcohol, and triphenylmethanol led to the corresponding C3-benzylic and allylic products 3s–v (52–95%).
 |
| | Scheme 3 Scope of indoles 1 and alcohols 2. Reaction conditions: 1 (1 mmol), PtCl2(PhCN)2 (0.05 mmol), 2 (1.2 mmol), and 1,2-dichloroethane (4 mL). | |
Hammett study
To demonstrate the electronic effect of the substituents on the rates of the sp3 C–O bond cleavage of alcohols, the relative rates of the coupling of 4-substituted benzhydrols 2X with 5-nitroindole (1a) were examined (see ESI, Scheme S2†).23Fig. 4 shows good correlation (R2 = 0.99) between the log(kX/kH) and the σp values of the respective substituents. The resulting negative ρ value of 2.9 indicates that there is a build-up of positive charge in the transition state.
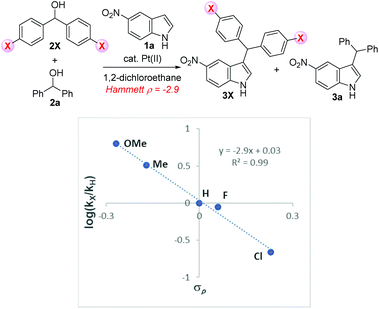 |
| | Fig. 4 Hammett plots for the rate constants of benzylation by various substituted 4-benzhydrols 2X (X = OMe, Me, F and Cl groups). | |
Control experiments
1H NMR study was performed to ascertain the role of the Pt(II) catalyst (Scheme 4A). Roy et al. reported that all proton signals of the C3-palladated indole complex were shifted downfield from those of free indole, except for the C3-H signal.7c In contrast, when 5-nitroindole (1a) was treated with PtCl2(PhCN)2 (1 equiv.) in CDCl3 at 60 °C, the 1H NMR spectrum was unchanged from that of 1a. These results suggested that the Pt(II) catalyst did not interact with the electron-deficient indole 1a due to the low Lewis basicity of 1a. Interestingly, the water-promoted dehydrative coupling of 5-cyanoindole (1b) with alcohol 2a proceeded smoothly using our catalytic system, whereas the use of the PdCl2(MeCN)2 catalyst was less effective, clearly demonstrating the superiority of the Pt(II) catalytic system for the C3-benzylation of 1b (Scheme 4B).
 |
| | Scheme 4 Control experiments. | |
Mechanistic considerations
On the basis of these results and our previous report, we propose a catalytic system for the water-promoted dehydrative coupling of 4-nitroindole (1a) with benzhydrol (2a) catalyzed by Pt(II) as illustrated in Scheme 5. Initially, a ligand exchange of PtCl2(PhCN)2 with alcohol 2a generates intermediate A (step 1: ligand exchange), and sp3 C–O bond activation occurs to generate a benzylic cation B (step 2: C–O bond cleavage). The observed negative Hammett ρ value of 2.9 in the reaction of para-substituted benzhydryl alcohols 2 clearly shows cationic charge build up during the rate-determining sp3 C–O bond cleavage (see Fig. 4). Additionally, the direct substitution of trityl alcohol affords the corresponding desired product 3u, which is in agreement with the reaction proceeding through an SN1 reaction. Notably, the presence of a few water molecules leads to an acceleration of the dehydrative reaction rate, suggesting that the transition state TS was stabilized by water molecules. Next, the hydroxyl anion of platinum species C acts as a base to remove the acidic proton of substrate 1a, which attacks the electrophilically active benzyl cation B to afford the corresponding C3-benzylated product 3a (step 3: C3-benzylation).
 |
| | Scheme 5 Proposed mechanism. | |
Scale-up experiment
Finally, we examined the scalability of our catalytic system (Scheme 6). To demonstrate the utility of our efficient and simple protocol, a gram scale reaction of 1a in the presence of only 1 mol% of the Pt(II) catalyst with 1 equiv. of water was carried out. The dehydrative coupling reaction of 1a with 2a was completed in 36 h. After cooling, n-hexane was added to the reaction mixture. The precipitate was filtered to give the desired product 3a in 92% isolated yield. Notably, the developed process avoids using column chromatography. To the best of our knowledge, this is the first example of a direct dehydrative coupling reaction utilizing a Pt(II) catalyst with an additional molecule of water.
 |
| | Scheme 6 Scale-up experiment. | |
Conclusions
We have reported an efficient new protocol for the Pt(II)-catalyzed dehydrative C–C bond formation reaction to construct 3-benzylindoles. Notably, water molecules were found to be crucial as a cocatalyst for the success of our catalytic system. Our proposed strategy was applicable to the direct C3-benzylation of electron-deficient indoles. This simple protocol can be performed under mild conditions in an atom-economical process without the need for bases or other additives, furnishing the desired products in moderate to excellent yields along with only stoichiometric amounts of water as the sole co-product.
Experimental
All of the starting materials and solvents were purchased from Sigma-Aldrich Japan (Tokyo, Japan), FUJIFILM Wako Pure Chemical Co. (Osaka, Japan), and TCI Co., Ltd (Tokyo, Japan). All commercially available reagents and solvents were used without further purification. CHROMATOREX Q-PACK SI50 (Fuji Silysia Chemical Ltd, Japan) was used for flash column chromatography. All melting points were determined using a Yanako micro melting point apparatus without correction. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a JEOL ECS400 spectrometer. IR spectra were measured with a JASCO FT/IR-4100 spectrometer. Mass spectra were obtained using a JEOL JMS-700 MStation Mass Spectrometer.
General procedure I
A mixture of indoles 1 (0.5 mmol), PtCl2(PhCN)2 (11.6 mg, 0.025 mmol) and alcohols 2 (0.6 mmol) in 1,2-dichloroethane (2 mL) was heated at 60 or 90 °C under air. After cooling, the reaction mixture was poured into water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, n-hexane/EtOAc) to give the desired product 3.
General procedure II
A mixture of indoles 1 (1 mmol), PtCl2(PhCN)2 (23 mg, 0.05 mmol) and alcohols 2 (1.2 mmol) in 1,2-dichloroethane (4 mL) was heated at 90 °C for 17 h under air. After cooling, the reaction mixture was poured into water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, n-hexane/EtOAc) to give the desired product 3.
3-Benzhydryl-5-nitro-1H-indole 3a.
Following the general procedure II, 3a was obtained as a yellow solid; yield 315 mg (96%); mp 275–278 °C [257–265 °C, lit];9b IR (KBr) (cm−1) 3304, 3025, 1625, 1500; 1H NMR (400 MHz, CDCl3): δ 5.71 (s, 1H), 6.76 (dd, J = 2.1, 0.9 Hz, 1H), 7.19–7.33 (m, 10H), 7.39 (d, J = 8.5 Hz, 1H), 8.09 (ddd, J = 9.0, 2.1, 0.9 Hz, 1H), 8.20 (dd, J = 1.4, 0.7 Hz, 1H), 8.32 (br, 1H); 13C NMR (100 MHz, CDCl3): δ 48.4, 111.1, 117.1, 118.0, 122.7, 126.4, 126.7, 126.9, 128.5, 128.8, 139.6, 141.7, 142.9; MS (FAB): m/z 329 [M + H]+.
3-Benzhydryl-4-nitro-1H-indole 3b.
Following the general procedure I, 3b was obtained as a brown solid; yield 131 mg (80%); Rf = 0.5 (hexane/ethyl acetate = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); mp 267–269 °C; IR (KBr) (cm−1) 3361, 3021, 2339, 1510; 1H NMR (400 MHz, CDCl3): δ 6.04 (s, 1H), 6.65 (dd, J = 2.8, 1.1 Hz, 1H), 7.08–7.13 (m, 4H), 7.16–7.29 (m, 7H), 7.62 (ddd, J = 7.9, 5.8, 0.9 Hz, 2H), 8.37 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 49.3, 116.7, 117.4, 118.4, 119.9, 121.1, 126.3, 128.3, 128.8, 129.1, 139.1, 143.9, 144.1; MS (EI): m/z (%) 328 (M+, 21), 280 (100); HRMS (EI): m/z [M+] calcd for C21H16N2O2 328.1208, found 328.1213.
:1); mp 267–269 °C; IR (KBr) (cm−1) 3361, 3021, 2339, 1510; 1H NMR (400 MHz, CDCl3): δ 6.04 (s, 1H), 6.65 (dd, J = 2.8, 1.1 Hz, 1H), 7.08–7.13 (m, 4H), 7.16–7.29 (m, 7H), 7.62 (ddd, J = 7.9, 5.8, 0.9 Hz, 2H), 8.37 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 49.3, 116.7, 117.4, 118.4, 119.9, 121.1, 126.3, 128.3, 128.8, 129.1, 139.1, 143.9, 144.1; MS (EI): m/z (%) 328 (M+, 21), 280 (100); HRMS (EI): m/z [M+] calcd for C21H16N2O2 328.1208, found 328.1213.
3-Benzhydryl-6-nitro-1H-indole 3c.
Following the general procedure I, 3c was obtained as a yellow solid; yield 123 mg (75%); mp 186–188 °C [186–190 °C, lit];9b IR (KBr) (cm−1) 3328, 3025, 1587, 1496; 1H NMR (400 MHz, CDCl3): δ 5.67 (s, 1H), 6.89 (dd, J = 2.5, 0.9 Hz, 1H), 7.19–7.33 (m, 11H), 7.88 (dd, J = 8.9, 2.1 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 8.42 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 48.5, 108.1, 115.0, 120.0, 121.1, 126.7, 128.5, 128.8, 129.7, 131.5, 135.1, 143.0, 143.4; MS (FAB): m/z 329 [M + H]+.
3-Benzhydryl-7-nitro-1H-indole 3d.
Following the general procedure I, 3d was obtained as a yellow solid; yield 156 mg (95%); mp 194–197 °C [190–194 °C, lit];9b IR (KBr) (cm−1) 3369, 3032, 1517, 1492; 1H NMR (400 MHz, CDCl3): δ 5.68 (s, 1H), 6.77 (dd, J = 2.3, 1.1 Hz, 1H), 7.06 (t, J = 7.9 Hz, 1H), 7.19–7.34 (m, 10H), 7.54 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 8.0 Hz, 1H), 9.72 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 48.5, 118.9, 119.4, 121.5, 126.4, 126.6, 128.1, 128.5, 128.9, 130.2, 130.8, 132.9, 143.0; MS (FAB): m/z 329 [M + H]+.
3-Benzhydryl-1H-indole-5-carboxylic acid 3e.
Following the general procedure I, 3e was obtained as a white solid; yield 131 mg (80%); Rf = 0.75 (CHCl3/MeOH = 4:1); mp 228–233 °C; IR (KBr) (cm−1) 3430, 3025, 1683, 1619; 1H NMR (400 MHz, CDCl3): δ 5.73 (s, 1H), 6.67 (dd, J = 2.2, 1.1 Hz, 1H), 7.19–7.32 (m, 12H), 7.39 (dd, J = 8.6, 0.5 Hz, 1H), 7.93 (dd, J = 8.5, 1.6 Hz, 1H), 8.10 (t, J = 0.8, 1H), 8.17 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 48.4, 110.8, 120.6, 121.7, 123.7, 124.1, 125.3, 126.4, 126.7, 128.4, 128.9, 139.7, 143.5, 172.4; MS (FAB): m/z 328 [M + H]+; anal. calcd for C22H17NO2: C, 80.71; H, 5.23; N, 4.28. Found: C, 80.44; H, 5.25; N, 4.29.
3-Benzhydryl-1H-indole-6-carboxylic acid 3f.
Following the general procedure II, 3f was obtained as a white solid; yield 261 mg (79%); Rf = 0.75 (CHCl3/MeOH = 4:1); mp >300 °C; IR (KBr) (cm−1) 3373, 3026, 2875, 1661; 1H NMR (400 MHz, DMSO-d6): δ 5.72 (s, 1H), 6.95 (dd, J = 2.5, 0.7 Hz, 1H), 7.13–7.34 (m, 11H), 7.47 (dd, J = 8.4, 1.5 Hz, 1H), 8.01 (dd, J = 1.4, 0.7 Hz, 1H), 11.29 (d, J = 2.1 Hz, 1H), 12.48 (s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 47.7, 113.6, 118.1, 118.5, 119.2, 123.2, 126.1, 127.6, 128.2, 128.4, 129.5, 135.8, 143.9, 168.2; MS (EI): m/z (%) 327 (M+, 100); HRMS (EI): m/z [M+] calcd for C22H17NO2 327.1255, found 327.1258.
3-Benzhydryl-1H-indole-7-carboxylic acid 3g.
Following the general procedure II, 3g was obtained as a pale brown solid; yield 225 mg (69%); Rf = 0.33 (hexane/ethyl acetate = 1:2); mp 208–212 °C; IR (KBr) (cm−1) 3453, 3032, 2538, 1682; 1H NMR (400 MHz, CDCl3): δ 5.70 (s, 1H), 6.71 (dd, J = 2.0, 0.9 Hz, 1H), 7.06 (t, J = 7.7 Hz, 1H), 7.19–7.33 (m, 11H), 7.49 (d, J = 7.8 Hz, 1H), 7.96 (dd, J = 7.6, 0.9 Hz, 1H), 9.58 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 48.7, 111.4, 118.9, 120.2, 125.2, 125.6, 126.4, 126.8, 128.3, 128.4, 128.9, 136.9, 143.6, 172.9; MS (EI): m/z (%) 327 (M+, 13), 59 (100); HRMS (FAB): m/z [M+] calcd for C22H17NO2 327.1255, found 327.1260.
3-Benzhydryl-1H-indole-5-carbonitrile 3h.
Following the general procedure II, 3h was obtained as a white solid; yield 163 mg (53%); mp 223–227 °C [226–229 °C, lit];9b IR (KBr) (cm−1) 3330, 3026, 2218, 1615; 1H NMR (400 MHz, CDCl3): δ 5.64 (s, 1H), 6.71 (dd, J = 2.4, 1.0 Hz, 1H), 7.17–7.34 (m, 10H), 7.40 (s, 2H), 7.55 (s, 1H), 8.26 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 48.6, 102.5, 112.0, 120.7, 121.0, 125.2, 125.6, 126.1, 126.7, 126.8, 128.5, 128.8, 138.4, 143.0; MS (FAB): m/z 309 [M + H]+.
3-Benzhydryl-5-chloro-1H-indole 3i.
Following the general procedure II, 3i was obtained as an amorphous solid; yield 229 mg (72%); Rf = 0.14 (hexane/ethyl acetate = 2:1); IR (KBr) (cm−1) 3425, 3062, 3025, 2869, 1657, 1594; 1H NMR (400 MHz, CDCl3): δ 5.61 (s, 1H), 6.59 (s, 1H), 7.07–7.34 (m, 13H), 7.97 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 48.6, 112.1, 119.3, 119.7, 122.5, 125.1, 125.4, 126.4, 128.1, 128.4, 128.9, 135.0, 143.5; MS (FAB): m/z 319 [M + 2]+, 317 [M]+; HRMS (FAB): m/z [M+] calcd for C21H16ClN 317.0968, found 317.0972.
3-Benzhydryl-1H-indole-2-carboxylic acid 3j.
Following the general procedure II, 3j was obtained as a white solid; yield 241 mg (74%); mp 246–248 °C [245–248 °C, lit];9b IR (KBr) (cm−1) 3410, 3028, 2575, 1670; 1H NMR (400 MHz, CDCl3): δ 6.74 (s, 1H), 6.90 (ddd, J = 8.5, 7.1, 0.9 Hz, 1H), 7.03 (dd, J = 8.2, 0.7 Hz, 1H), 7.18–7.31 (m, 12H), 7.34 (d, J = 8.2, 1H), 8.88 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 47.1, 112.0, 120.5, 122.6, 123.7, 125.9, 126.3, 127.3, 128.2, 128.4, 129.3, 136.7, 143.1, 166.5; MS (FAB): m/z 328 [M + H]+.
3-Benzhydryl-1-methyl-2-phenyl-1H-indole 3k.
Following the general procedure I, 3k was obtained as a white solid; yield 121 mg (65%); Rf = 0.64 (hexane/ethyl acetate = 8:1); mp 165–168 °C; IR (KBr) (cm−1) 3059, 2937, 1598; 1H NMR (400 MHz, CDCl3): δ 3.59 (s, 3H), 5.53 (s, 1H), 6.93 (ddd, J = 8.0, 7.1, 0.9 Hz, 1H), 7.10–7.35 (m, 15H), 7.39–7.45 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 30.9, 48.1, 109.3, 115.2, 119.2, 121.2, 121.4, 125.9, 126.9, 128.0, 128.3, 128.3, 129.1, 130.8, 131.9, 137.4, 139.0, 144.4; MS (FAB): m/z 373 [M]+, HRMS (FAB): m/z [M+] calcd for C28H23N 373.1825, found 373.1831.
3-[Bis(4-methoxyphenyl)methyl]-5-nitro-1H-indole 3l9a.
Following the general procedure I, 3l was obtained as a brown solid; yield 165 mg (86%); mp 226–231 °C; IR (KBr) (cm−1) 3288, 2831, 1619, 1508; 1H NMR (400 MHz, CDCl3): δ 3.79 (s, 6H), 5.60 (s, 1H), 6.74 (d, J = 2.1 Hz, 1H), 6.83 (d, J = 8.7 Hz, 4H), 7.11 (d, J = 8.7 Hz, 4H), 7.34 (d, J = 8.9 Hz, 1H), 8.08 (dd, J = 8.9, 2.3 Hz, 1H), 8.21 (d, J = 2.1 Hz, 1H), 8.30 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 46.7, 55.3, 111.0, 113.9, 117.2, 117.9, 123.4, 126.4, 126.8, 129.8, 136.5, 139.7, 141.6, 158.2; MS (FAB): m/z 389 [M + H]+.
3-[Bis(4-chlorophenyl)methyl]-5-nitro-1H-indole 3m.
Following the general procedure I, 3m was obtained as a yellow solid; yield 194 mg (98%); Rf = 0.37 (hexane/ethyl acetate = 2:1); mp 285–289 °C; IR (KBr) (cm−1) 3287, 1489; 1H NMR (400 MHz, CDCl3): δ 5.66 (s, 1H), 6.73 Hz, (1H), 7.11 (d, J = 8.5 Hz, 4H), 7.24–7.33 (m, 4H), 7.42 (d, J = 9.2 Hz, 1H), 8.11 (dd, J = 8.9, 2.1 Hz, 1H), 8.18 (d, J = 1.8 Hz, 1H), 8.38 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 47.1, 111.3, 116.9, 118.3, 121.7, 126.1, 126.9, 128.8, 130.1, 132.8, 139.6, 141.0, 141.8; MS (EI): m/z (%) 400 (M+ + 4, 10), 398 (M+ + 2, 59), 396 (M+, 89), 366 (100); HRMS (EI): m/z [M+] calcd for C21H14Cl2N2O2 396.0430, found 396.0431.
3-[Bis(4-fluorophenyl)methyl]-5-nitro-1H-indole 3n.
Following the general procedure I, 3n was obtained as a yellow solid; yield 178 mg (98%); Rf = 0.34 (hexane/ethyl acetate = 2:1); mp 264–270 °C; IR (KBr) (cm−1) 3288, 1606, 1506; 1H NMR (400 MHz, CDCl3): δ 5.68 (s, 1H), 6.72 (t, J = 1.1 Hz, 1H), 7.00 (t, J = 8.0 Hz, 4H), 7.15 (dd, J = 8.4, 5.3 Hz, 4H), 7.41 (d, J = 9.4 Hz, 1H), 8.11 (dd, J = 9.0, 1.4 Hz, 1H), 8.18 (d, J = 2.1 Hz, 1H), 8.37 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 46.9, 111.2, 115.5 (JCF = 22.0 Hz), 116.9, 118.2, 122.4, 126.2, 126.8, 130.2 (JCF = 8.6 Hz), 138.5 (JCF = 2.9 Hz), 139.7, 141.8, 161.7 (JCF = 245.4 Hz); MS (FAB): m/z 365 [M + H]+; HRMS (FAB): m/z [M + H]+ calcd for C21H14F2N2O2 365.1098, found 365.1101.
3-[(4-Methoxyphenyl)(phenyl)methyl]-5-nitro-1H-indole 3o.
Following the general procedure I, 3o was obtained as a yellow solid; yield 147 mg (82%); Rf = 0.31 (hexane/ethyl acetate = 2:1); mp 214–218 °C; IR (KBr) (cm−1) 3271, 3025, 2837, 2359, 1624; 1H NMR (400 MHz, CDCl3): δ 3.79 (s, 3H), 5.66 (s, 1H), 6.75 (t, J = 1.1 Hz, 1H), 6.84 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.7 Hz, 2H), 7.17–7.33 (m, 5H), 7.38 (d, J = 8.9 Hz, 1H), 8.09 (dd, J = 8.8, 2.1 Hz, 1H), 8.20 (d, J = 1.8 Hz, 1H), 8.32 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 47.6, 55.3, 111.0, 113.9, 117.2, 118.0, 123.0, 126.4, 126.6, 126.8, 128.5, 128.7, 129.8, 135.1, 139.7, 141.6, 143.3, 158.3; MS (EI): m/z (%) 358 (M+, 100); HRMS (EI): m/z [M+] calcd for C22H18N2O3 358.1313, found 358.1318.
5-Nitro-3-[phenyl(p-tolyl)methyl]-1H-indole 3p.
Following the general procedure I, 3p was obtained as a yellow solid; yield 158 mg (92%); Rf = 0.4 (hexane/ethyl acetate = 2:1); mp 265–270 °C; IR (KBr) (cm−1) 3296, 3025, 1509; 1H NMR (400 MHz, CDCl3): δ 2.33 (s, 3H), 5.67 (s, 1H), 6.76 (t, J = 1.1 Hz, 1H), 7.10 (s, 4H), 7.19–7.33 (m, 5H), 7.38 (d, J = 8.9 Hz, 1H), 8.08 (dd, J = 8.9, 2.1 Hz, 1H), 8.20 (d, J = 1.8 Hz, 1H), 8.31 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 21.0, 48.0, 111.0, 117.2, 118.0, 122.8, 126.6, 126.8, 128.5, 128.7, 128.8, 129.2, 136.2, 139.6, 140.0, 141.7, 143.2; MS (EI): m/z (%) 342 (M+, 100); HRMS (EI): m/z [M+] calcd for C22H18N2O2 342.1364, found 342.1369.
3-[(4-Chlorophenyl)(phenyl)methyl]-5-nitro-1H-indole 3q.
Following the general procedure I, 3q was obtained as a yellow solid; yield 163 mg (90%); Rf = 0.4 (hexane/ethyl acetate = 2:1); mp 259–261 °C; IR (KBr) (cm−1) 3306, 1489; 1H NMR (400 MHz, CDCl3): δ 5.68 (s, 1H), 6.74 (t, J = 1.1 Hz, 1H), 7.11–7.21 (m, 4H), 7.23–7.35 (m, 5H), 7.40 (d, J = 8.9 Hz, 1H), 8.10 (dd, J = 9.4, 2.1 Hz, 1H), 8.19 (d, J = 2.1 Hz, 1H), 8.36 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 47.8, 111.2, 117.0, 118.1, 122.1, 126.3, 126.9, 128.7, 128.7, 128.7, 130.2, 132.5, 139.6, 141.5, 141.8, 142.4; MS (EI): m/z (%) 364 (M+ + 2, 34), 362 (M+, 100), HRMS (EI): m/z [M+] calcd for C21H15ClN2O2 362.0819, found 362.0823.
5-Nitro-3-{phenyl[3-(trifluoromethyl)phenyl]methyl}-1H-indole 3r.
Following the general procedure I (the reaction was conducted at 90 °C), 3r was obtained as a yellow solid; yield 143 mg (72%); Rf = 0.4 (hexane/ethyl acetate = 2:1); mp 206–208 °C; IR (KBr) (cm−1) 3310, 1625; 1H NMR (400 MHz, DMSO-d6): δ 6.06 (s, 1H), 7.06 (d, J = 2.3 Hz, 1H), 7.20–7.40 (m, 5H), 7.50–7.65 (m, 5H), 7.98 (dd, J = 9.2, 1.8 Hz, 1H), 8.14 (d, J = 2.3 Hz, 1H), 11.8 (brs, 1H); 13C NMR (100 MHz, DMSO-d6): δ 47.3, 112.8, 116.4, 117.3, 120.3, 123.8 (q, JCF = 3.8 Hz), 124.8 (q, JCF = 272.2 Hz), 125.4 (q, JCF = 3.8 Hz), 126.2, 127.2, 128.8, 129.0, 129.2, 129.6 (q, JCF = 31.6 Hz), 130.1, 133.2, 140.3, 140.8, 143.4, 145.7; MS (FAB): m/z 397 [M + H]+; anal. calcd for C22H15F3N2O2: C, 66.67; H, 3.81; N, 7.07. Found: C, 66.45; H, 3.89; N, 6.97.
3-[1-(4-Methoxyphenyl)ethyl]-5-nitro-1H-indole 3s.
Following the general procedure II, 3s was obtained as a yellow solid; yield 268 mg (90%); Rf = 0.29 (hexane/ethyl acetate = 2:1); mp 188–191 °C; IR (KBr) (cm−1) 3308, 2956, 1623; 1H NMR (400 MHz, CDCl3): δ 1.69 (d, J = 7.1 Hz, 3H), 3.78 (s, 3H), 4.36 (q, J = 7.1 Hz, 1H), 6.84 (d, J = 8.7 Hz, 2H), 7.15 (dd, J = 2.3, 0.9 Hz, 1H), 7.19 (d, J = 8.7 Hz, 2H), 7.36 (d, J = 8.9 Hz, 1H), 8.07 (dd, J = 8.9, 2.1 Hz, 1H), 8.33 (brs, 1H), 8.34 (d, J = 2.1 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 22.5, 35.9, 55.3, 111.0, 113.9, 117.1, 117.8, 123.8, 124.6, 126.3, 128.2, 137.8, 139.6, 141.5, 158.1; MS (FAB): m/z (%) 297 [M + H]+, HRMS (FAB): m/z [M + H]+ calcd for C17H16N2O3 297.1235, found 297.1239.
5-Nitro-3-(1-phenylethyl)-1H-indole 3t.
Following the general procedure II, 3t was obtained as a yellow solid; yield 138 mg (52%); Rf = 0.45 (hexane/ethyl acetate = 2:1); mp 137–144 °C; IR (KBr) (cm−1) 3297, 3025, 2869, 1625; 1H NMR (400 MHz, CDCl3): δ 1.72 (d, J = 7.1 Hz, 3H), 4.40 (q, J = 7.1 Hz, 1H), 7.15–7.33 (m, 6H), 7.36 (d, J = 8.9 Hz, 1H), 8.07 (dd, J = 8.9, 2.3 Hz, 1H), 8.33 (d, J = 2.1 Hz, 1H), 8.35 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 22.3, 36.7, 111.0, 117.0, 117.8, 124.0, 124.2, 126.3, 126.4, 127.3, 128.6, 139.6, 141.5, 145.7; MS (FAB): m/z (%) 267 [M + H]+, HRMS (FAB): m/z [M + H]+ calcd for C16H14N2O2 267.1130, found 267.1140.
(E)-3-(1,3-Diphenylallyl)-5-nitro-1H-indole 3u24.
Following the general procedure I, 3u was obtained as a yellow solid; yield 161 mg (90%); mp 181–185 °C; IR (KBr) (cm−1) 3304, 3025, 2357, 1514; 1H NMR (400 MHz, CDCl3): δ 5.16 (d, J = 7.3 Hz, 1H), 6.43 (d, J = 15.7 Hz, 1H), 6.70 (dd, J = 15.9, 7.1 Hz, 1H), 7.09 (dd, J = 2.3, 0.9 Hz, 1H), 7.19–7.42 (m, 11H), 8.10 (dd, J = 9.0, 2.3 Hz, 1H), 8.38 (brs, 1H), 8.39 (d, J = 2.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 45.8, 111.1, 117.1, 118.0, 121.5, 125.6, 126.2, 126.4, 126.9, 127.5, 128.3, 128.6, 128.7, 131.3, 131.5, 137.1, 139.6, 141.7, 142.4; MS (FAB): m/z 355 [M + H]+.
5-Nitro-3-trityl-1H-indole 3v.
Following the general procedure I, 3u was obtained as a yellow solid; yield 192 mg (95%); mp >300 °C [298 °C, lit];25 IR (KBr) (cm−1) 3401, 3057, 1507; 1H NMR (400 MHz, CDCl3): δ 7.06 (d, J = 2.3 Hz, 1H), 7.16–7.30 (m, 15H), 7.36 (d, J = 8.9 Hz, 1H), 7.59 (d, J = 1.8 Hz, 1H), 8.01 (dd, J = 9.3, 2.3 Hz, 1H), 8.33 (brs, 1H); 13C NMR (100 MHz, CDCl3): δ 59.3, 111.1, 117.7, 119.7, 126.5, 127.2, 127.7, 128.0, 126.9, 130.5, 139.8, 141.5, 145.7; MS (FAB): m/z 405 [M + H]+.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the JSPS KAKENHI Grant Number 19K07003.
Notes and references
- For a review, see:
(a) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS;
(b) R. N. Butler and A. G. Coyne, Org. Biomol. Chem., 2016, 14, 9945–9960 RSC;
(c) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
-
(a) Z. Xu, X. Yu, X. Sang and D. Wang, Green Chem., 2018, 20, 2571–2577 RSC;
(b) S. Hajra, S. Singha Roy, S. M. Aziz and D. Das, Org. Lett., 2017, 19, 4082–4085 CrossRef CAS PubMed;
(c) R. S. Tukhvatshin, A. S. Kucherenko, Y. V. Nelyubina and S. G. Zlotin, ACS Catal., 2017, 7, 2981–2989 CrossRef CAS.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
-
(a) G. J. Roth, R. Binder, F. Colbatzky, C. Dallinger, R. Schlenker-Herceg, F. Hilberg, S.-L. Wollin and R. Kaiser, J. Med. Chem., 2015, 58, 1053–1063 CrossRef CAS PubMed;
(b) K. McKeage, Drugs, 2015, 75, 75–82 CrossRef CAS PubMed;
(c) J. C. McKew, F. Lovering, J. D. Clark, J. Bemis, Y. Xiang, M. Shen, W. Zhang, J. C. Alvarez and D. Joseph-McCarthy, Bioorg. Med. Chem. Lett., 2003, 13, 4501–4504 CrossRef CAS PubMed;
(d) D. J. Rawson, K. N. Dack, R. P. Dickinson and K. James, Bioorg. Med. Chem. Lett., 2002, 12, 125–128 CrossRef CAS PubMed;
(e) R. T. Jacobs, P. R. Bernstein, L. A. Cronk, E. P. Vacek, L. F. Newcomb, D. Aharony, C. K. Buckner and E. J. Kusner, J. Med. Chem., 1994, 37, 1282–1297 CrossRef CAS PubMed.
- For a review, see:
(a) M. Dryzhakov, E. Richmond and J. Moran, Synthesis, 2016, 935–959 CAS;
(b) R. Kumar and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 1121–1146 RSC;
(c) E. Emer, R. Sinisi, M. G. Capdevila, D. Petruzziello, F. De Vincentiis and P. G. Cozzi, Eur. J. Org. Chem., 2011, 647–666 CrossRef CAS.
-
(a) G. Di Gregorio, M. Mari, F. Bartoccini and G. Piersanti, J. Org. Chem., 2017, 82, 8769–8775 CrossRef CAS PubMed;
(b) M.-H. Zhuo, G.-F. Liu, S.-L. Song, D. An, J. Gao, L. Zheng and S. Zhang, Adv. Synth. Catal., 2016, 358, 808–815 CrossRef CAS;
(c) D. Das and S. Roy, Adv. Synth. Catal., 2013, 355, 1308–1314 CrossRef CAS;
(d) Y.-L. Liu, L. Liu, Y.-L. Wang, Y.-C. Han, D. Wang and Y.-J. Chen, Green Chem., 2008, 10, 635–640 RSC.
-
(a) H. Hikawa, F. Kotaki, S. Kikkawa and I. Azumaya, J. Org. Chem., 2019, 84, 1972–1979 CrossRef CAS PubMed;
(b) H. Hikawa, H. Imamura, S. Kikkawa and I. Azumaya, Green Chem., 2018, 20, 3044–3049 RSC;
(c) H. Hikawa, R. Ichinose, S. Kikkawa and I. Azumaya, Green Chem., 2018, 20, 1297–1305 RSC;
(d) H. Hikawa, H. Suzuki and I. Azumaya, J. Org. Chem., 2013, 78, 12128–12135 CrossRef CAS PubMed.
- For the dehydrative C–C bond formation of 5-nitroindole with benzhydrol, see:
(a) A. Zhu, W. Feng, L. Li, Q. Li and J. Wang, Catal. Lett., 2017, 147, 261–268 CrossRef CAS;
(b) H. Hikawa, Y. Machino, M. Toyomoto, S. Kikkawa and I. Azumaya, Org. Biomol. Chem., 2016, 14, 7038–7045 RSC;
(c) J. S. Yadav, B. V. S. Reddy and A. S. Reddy, J. Mol. Catal. A: Chem., 2008, 280, 219–223 CrossRef CAS.
-
(a) P. Safar, S. Marchalin, M. Soral, J. Moncol and A. Daich, Org. Lett., 2017, 19, 4742–4745 CrossRef CAS PubMed;
(b) B. D. Mokar and R.-S. Liu, Chem. Commun., 2014, 50, 8966–8969 RSC;
(c) Y. Kwon, I. Kim and S. Kim, Org. Lett., 2014, 16, 4936–4938 CrossRef CAS PubMed.
- The smooth formation of a water-soluble PdCl2(H2O)2 complex from the PdCl2(MeCN)2 catalyst by loss of a CH3CN ligand was reported. See: S. Ganesamoorthy, K. Shanmugasundaram and R. Karvembu, J. Mol. Catal. A: Chem., 2013, 371, 118–124 CrossRef CAS.
- H. T. Dang, H. W. Lee, J. Lee, H. Choo, S. H. Hong, M. Cheong and H. Lee, ACS Catal., 2018, 8, 11854–11862 CrossRef CAS.
-
(a) D. Hueber, M. Teci, E. Brenner, D. Matt, J.-M. Weibel, P. Pale and A. Blanc, Adv. Synth. Catal., 2018, 360, 2453–2459 CrossRef CAS;
(b) J.-Q. Zhao, X.-J. Zhou, Y.-Z. Chen, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Adv. Synth. Catal., 2018, 360, 2482–2487 CrossRef CAS;
(c) U. Nath, D. Chowdhury and S. C. Pan, Adv. Synth. Catal., 2018, 360, 1628–1633 CrossRef CAS.
-
(a) Y.-S. Lee, Y.-H. Cho, S.-J. Lee, J.-K. Bin, J.-H. Yang, G.-S. Chae and C.-H. Cheon, Tetrahedron, 2015, 71, 532–538 CrossRef CAS;
(b) Y. Qiu, C. Fu, X. Zhang and S. Ma, Chem. – Eur. J., 2014, 20, 10314–10322 CrossRef CAS PubMed.
- For addition of water-promoted organic reactions, see:
(a) P. Xie, J. Wang, Y. Liu, J. Fan, X. Wo, W. Fu, Z. Sun and T.-P. Loh, Nat. Commun., 2018, 9, 1321 CrossRef PubMed;
(b) X. Yang, W. Yang, Y. Yao, Y. Deng, X. Zuo and D. Yang, J. Org. Chem., 2018, 83, 10097–10106 CrossRef CAS PubMed;
(c) W. Chen, W. Yang, R. Wu and D. Yang, Green Chem., 2018, 20, 2512–2518 RSC.
- The result showed that the rate constants for the reaction of 2a in the presence of H2O versus D2O were found to be 0.3501 h−1 and 0.2353 h−1, respectively, yielding kH/kD = 1.5.
-
(a) K. D. Beare and C. S. P. McErlean, Org. Biomol. Chem., 2013, 11, 2452–2459 RSC;
(b) S. Mellouli, L. Bousekkine, A. B. Theberge and W. T. S. Huck, Angew. Chem., Int. Ed., 2012, 51, 7981–7984 CrossRef CAS PubMed;
(c) J. K. Beattie, C. S. P. McErlean and C. B. W. Phippen, Chem. – Eur. J., 2010, 16, 8972–8974 CrossRef CAS PubMed.
-
(a) P. Vidossich, G. Ujaque and A. Lledos, Chem. Commun., 2012, 48, 1979–1981 RSC;
(b) H. Zhu and T. Ziegler, J. Organomet. Chem., 2006, 691, 4486–4497 CrossRef CAS;
(c) P. E. M. Siegbahn and R. H. Crabtree, J. Am. Chem. Soc., 1996, 118, 4442–4450 CrossRef CAS.
- R. J. Deeth and L. I. Elding, Inorg. Chem., 1996, 35, 5019–5026 CrossRef CAS PubMed.
- H. Kinoshita, H. Shinokubo and K. Oshima, Org. Lett., 2004, 6, 4085–4088 CrossRef CAS PubMed.
- P. G. Cozzi and L. Zoli, Angew. Chem., Int. Ed., 2008, 47, 4162–4166 CrossRef CAS PubMed.
-
(a) A. Melchior, M. Tolazzi, J. M. Martinez, R. R. Pappalardo and E. S. Marcos, J. Chem. Theory Comput., 2015, 11, 1735–1744 CrossRef CAS PubMed;
(b) A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911–4920 CrossRef CAS PubMed;
(c) D. P. Bancroft, C. A. Lepre and S. J. Lippard, J. Am. Chem. Soc., 1990, 112, 6860–6871 CrossRef CAS.
-
(a) D. Ye, R. Huang, H. Zhu, L.-H. Zou and D. Wang, Org. Chem. Front., 2019, 6, 62–69 RSC;
(b) E. Tan, O. Quinonero, M. Elena de Orbe and A. M. Echavarren, ACS Catal., 2018, 8, 2166–2172 CrossRef CAS PubMed;
(c) X. Yu, D.-S. Wang, Z. Xu, B. Yang and D. Wang, Org. Chem. Front., 2017, 4, 1011–1018 RSC;
(d) S. R. Huq, S. Shi, R. Diao and M. Szostak, J. Org. Chem., 2017, 82, 6528–6540 CrossRef CAS PubMed;
(e) K. Yang, F. Zhou, Z. Kuang, G. Gao, T. G. Driver and Q. Song, Org. Lett., 2016, 18, 4088–4091 CrossRef CAS PubMed.
- Z. Liu, L. Liu, Z. Shafiq, Y.-C. Wu, D. Wang and Y.-J. Chen, Tetrahedron Lett., 2007, 48, 3963–3967 CrossRef CAS.
- W. Hanefeld and I. Hunz, Arch. Pharm., 1993, 326, 323–329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/c9qo00831d |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Yuuki
Matsuura
,
Shoko
Kikkawa
*,
Yuuki
Matsuura
,
Shoko
Kikkawa











