
Highly convergent total synthesis of (+)-anaferine and (−)-dihydrocuscohygrine†
Javier
Torres
,
Marcos
Escolano
,
Fernando
Rabasa-Alcañiz
,
Alvaro
Sanz-Vidal
,
María
Sánchez-Roselló
* and
Carlos
del Pozo
 *
*
Departamento de Química Orgánica, Universidad de Valencia, 46100 Burjassot, Spain. E-mail: carlos.pozo@uv.es
First published on 22nd July 2019
Abstract
A unified and highly convergent total synthesis of anaferine and dihydrocuscohygrine alkaloids has been devised, taking advantage of the dual role of N-sulfinyl amines as nucleophilic nitrogen sources and chiral auxiliaries. A bidirectional cross metathesis reaction followed by a double intramolecular aza-Michael reaction led us to create the whole skeleton of the natural products in a very simple manner.
Introduction
Anaferine (1) is a C2-symmetrical bis-piperidine alkaloid with the two piperidine rings anchored by a 2-propanone unit (Fig. 1). It was isolated in 1962 from Withania somnifera, a plant known for its sedative and anticancer properties.1 The interest on this natural product has been lately renewed due to its nAChR agonist activity2 and its properties as an inhibitor of GluN2B-containing NMDA receptors,3 both activities related to neurodegenerative diseases. Dihydrocuscohygrine (2), which contains a C2-symmetrical bis-pyrrolidine moiety (Fig. 1), was isolated from Erythroxylon coca,4 albeit there is no report regarding its biological activity.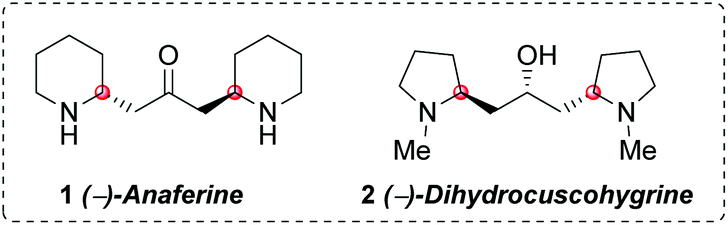 | ||
| Fig. 1 Anaferine and dihydrocuscohygrine alkaloids. | ||
Despite both alkaloids being characterized more than 30 years ago, an efficient asymmetric synthesis of their bis-piperidine and bis-pyrrolidine cores remains elusive. To date, only one stereoselective total synthesis of (−)-anaferine (1) has been reported in the literature.5 It is an eleven step-sequence starting from a chiral cycloheptene triol derivative (in turn obtained from tropone in five additional steps). The key step of this approach was a ruthenium-catalyzed tandem ring rearrangement metathesis reaction, which generated the carbon skeleton of the natural product. An analogous strategy was further extended to the synthesis of (+)-dihydrocuscohygrine (Scheme 1, eqn (1)).6 Both stereocenters present in the natural product are settled in the starting material.
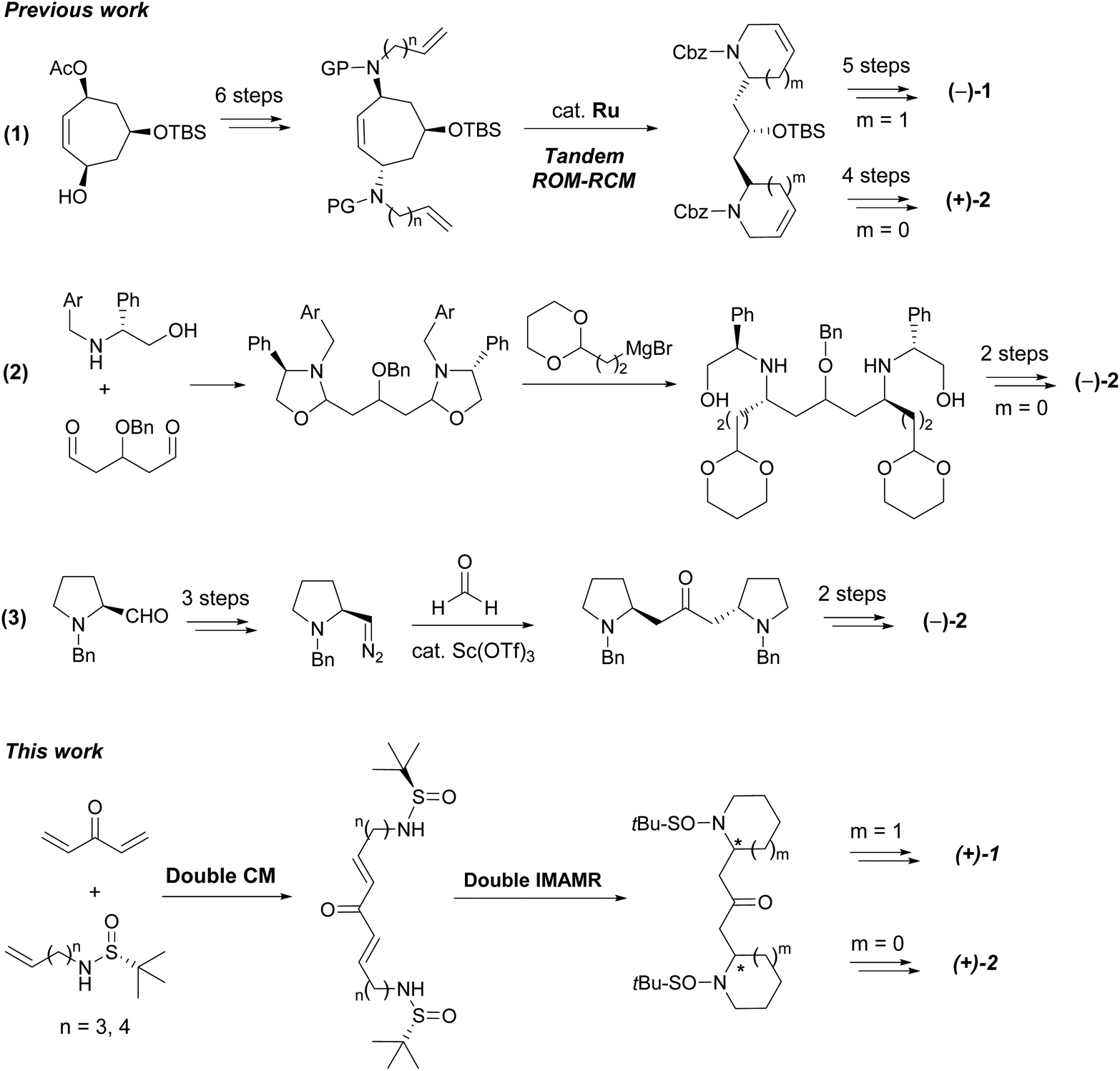 | ||
| Scheme 1 Synthesis strategies towards alkaloids 1 and 2. | ||
Two more syntheses of (−)-dihydrocuscohygrine (2) were also recently described. The first one employed a phenylglycinol derivative as the source of chirality. This was condensed with an appropriate dialdehyde compound to render the bis(1,3-oxazolidine). Further reaction with an acetal-containing Grignard derivative provided the corresponding diamine as a single diastereoisomer, which contains the stereocenters present in the final product. Reductive amination and chiral auxiliary release completed the total synthesis (Scheme 1, eqn (2)).7 The more recent total synthesis of (−)-dihydrocuscohygrine started from (S)-N-benzylprolinol which was converted into the corresponding diazo compound and then, it was subjected to a two-directional diazoalkane-carbonyl homologation reaction with formaldehyde (Scheme 1, eqn (3)).8 In this case, again the stereocenters present in the natural product arise from the starting material.
Results and discussion
N-Sulfinyl imines are among the most popular and efficient chiral auxiliaries employed in organic synthesis.9 After the addition of nucleophilic species (usually organometallic reagents), once the sulfinyl group has exerted its directing effect, it is typically removed and the free amino group is subjected to further transformations. However, the use of N-sulfinyl amines as nitrogen-centered nucleophiles is much less common.10 We have successfully applied this strategy to the total synthesis of the piperidine natural product pinidinol,11 as well as the azaphenalene natural products hyppodamine and epi-hyppodamine,12 using an intramolecular aza-Michael reaction as the key step,13 and the N-sulfinyl amine as an asymmetric nitrogen source. In the present paper, we describe a new application that provides a direct entry to bis-piperidine and bis-pyrrolidine skeletons taking advantage of the ability of N-sulfinyl amines to act as both chiral inducers and nucleophilic nitrogen sources. By using a bidirectional cross metathesis (CM) reaction and a double intramolecular aza-Michael reaction (IMAMR) as key steps, the whole skeleton of anaferine and dihydrocuscohygrine was constructed in a convergent manner (Scheme 1).14The first step of our study was the evaluation of the bidirectional CM reaction of sulfinyl amines 3 and divinyl ketone 4. Although the use of bis-enone 4 is troublesome due to its volatility and polymerization tendency, it could be prepared by the oxidation of divinyl alcohol 5 with DDQ in diethyl ether and used in solution without isolation.15 Then, sulfinyl amines 3 were added to this solution together with the 2nd generation Hoveyda–Grubbs catalyst (HG-II) and Ti(i-PrO)4 as a co-catalyst. After optimizing the reaction conditions, we found that the use of 3 and 4 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio in dichloromethane provided the best results after 16 h at room temperature. Thus, the bidirectional CM reaction took place efficiently, affording bis-enones 6 in good yields. The small amounts of single CM products 7 isolated were easily converted into the desired bis-enones 6 by treatment with the corresponding sulfonamides 3 under the same conditions (Scheme 2).
:2 ratio in dichloromethane provided the best results after 16 h at room temperature. Thus, the bidirectional CM reaction took place efficiently, affording bis-enones 6 in good yields. The small amounts of single CM products 7 isolated were easily converted into the desired bis-enones 6 by treatment with the corresponding sulfonamides 3 under the same conditions (Scheme 2).
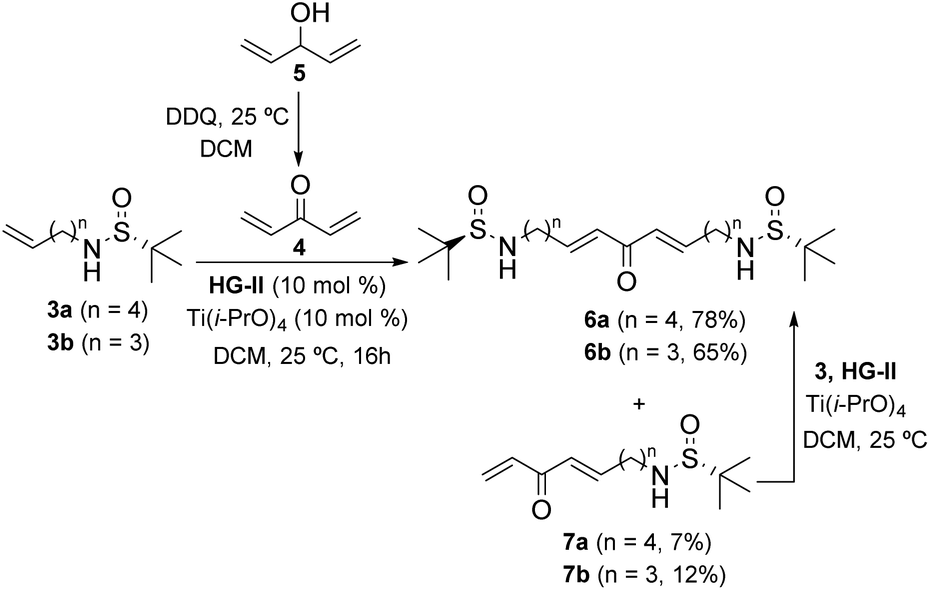 | ||
| Scheme 2 Bidirectional CM reaction between sulfinyl amines 3 and divinyl ketone 4. | ||
With substrates 6 in hand, we explored the feasibility of the double intramolecular aza-Michael reaction (IMAMR). This would be the key step of the synthesis since it would create the bis-pyrrolidine and bis-piperidine skeletons with the simultaneous generation of two stereocenters. In this context, we had previously studied the intramolecular aza-Michael reaction with N-sulfinyl amines,11 the final selectivity being clearly dependent on the base, the substrate and the size of the ring. In this scenario we tested several bases in order to find suitable conditions for the double cyclization. The results of our study on this reaction are summarized in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 6 | Base | T (°C) | Solvent | % yield (8 + 9) (dr 8:9)b,c |
| a Reactions were carried out with 6 (1 equiv.) and the base (2.2 equiv.) in the solvent and at the temperature indicated for 2 h. b Yields of compounds 8 and 9 after purification by flash column chromatography. c Diastereoisomers 8/9 were separated by column chromatography and obtained in the enantiomerically pure form. d No reaction. | |||||
| 1 | 6a | NaH | rt | THF | 98 (6:1) |
| 2 | 6a | NaH | 0 | THF | 85 (3:1) |
| 3 | 6a | NaH | rt | DCM | 87 (2.5:1) |
| 4 | 6a | NaH | rt | Toluene | 90 (2:1) |
| 5 | 6a | t-BuOK | rt | THF | 95 (1.5:1) |
| 6 | 6a | t-BuOK | −78 | THF | 93 (6:1) |
| 7 | 6a | KH | rt | THF | 97 (10:1) |
| 8 | 6a | DBU | rt | THF | —d |
| 9 | 6a | TBAF | rt | THF | —d |
| 10 | 6a | Cs2CO3 | rt | THF | —d |
| 11 | 6a | KHMDS | −78 | THF | 92 (5:1) |
| 12 | 6b | NaH | rt | THF | 90 (20:1) |
| 13 | 6b | NaH | 0 | THF | 87 (18:1) |
| 14 | 6b | t-BuOK | rt | THF | 95 (10:1) |
| 15 | 6b | t-BuOK | −78 | THF | 92 (8:1) |
| 16 | 6b | KHMDS | −78 | THF | 92 (8:1) |
| 17 | 6b | KH | rt | THF | —d |
| 18 | 6b | TBAF | rt | THF | —d |
| 19 | 6b | DBU | rt | THF | —d |
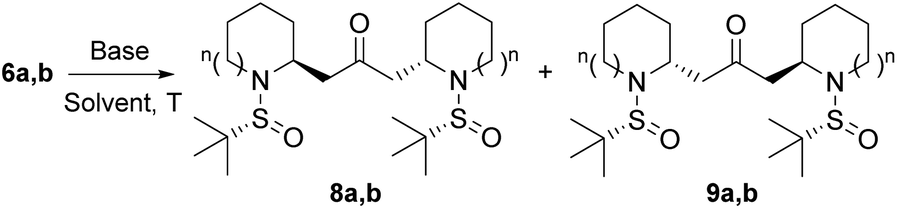
The first attempt to perform the double IMAMR was made on compound 6a with sodium hydride at room temperature. Under these conditions, a 6:1 mixture of diastereoisomers 8a and 9a was obtained in excellent 98% yield (Table 1, entry 1). When the temperature was reduced to 0 °C, the diastereoselectivity dropped to 3:1 (Table 1, entry 2). Other solvents such as dichloromethane and toluene were also tested, affording good yields but lower diastereoselectivity compared to the reaction in THF (Table 1, entries 3 and 4). Then, we explored other bases different from NaH to promote the double IMAMR. Thus, potassium tert-butoxide gave the desired bis-piperidines 8a/9a in very good yields (Table 1, entries 5 and 6) albeit only at −78 °C the diastereoselectivity was comparable to that obtained with NaH. The best results were achieved when KH was employed as the base, yielding compounds 8a/9a in a 10:1 ratio and excellent yield (97%) (Table 1, entry 7). Other bases previously employed in intramolecular aza-Michael reactions such as DBU,16 TBAF17 or the inorganic base Cs2CO3 failed to promote this reaction (Table 1, entries 8–10). Finally, a stronger base, namely potassium bis(trimethylsilyl)amide (KHMDS) did not improve the yield or selectivity (Table 1, entry 11). Since both diastereoisomers 8a and 9a were easily separated by column chromatography, conditions depicted in entry 7 (KH in THF at room temperature) were considered as the optimal conditions to obtain bis-piperidine 8a and continue the synthesis of anaferine.
We then extended the optimization process to substrate 6b. In this case, NaH provided the diasteroisomeric mixture of bis-pyrrolidines 8b/9b in 90% yield and an excellent 20:1 ratio at room temperature (Table 1, entry 12), while the reaction at 0 °C did not improve the yield or diastereoselectivity (Table 1, entry 13). The use of t-BuOK either at room temperature or at −78 °C afforded compounds 8b/9b in very good yield (95 an 92%, respectively) but with lower selectivity compared to that obtained with NaH (Table 1, entries 14 and 15). A similar result was observed when KHMDS was employed in THF at −78 °C (Table 1, entry 16). Other bases such as KH, DBU14 and TBAF15 were not effective to promote the desired transformation (Table 1, entries 17–19). Therefore, conditions indicated in entry 12 (NaH in THF at room temperature) were chosen as the best conditions to obtain bis-pyrrolidine 8b for the synthesis of dihydrocuscohygrine.
The results shown in Table 1 indicate that the selectivity in those cyclizations is clearly dependent on the size of the ring formed and the base employed. On the other hand, low temperatures affect negatively the diastereoselectivity providing the reactions at rt the best dr. The selectivity achieved could be explained on the basis of a double chair like transition state, where the nitrogen amide attacks the Si face of both sides of the bis-enone, yielding the final bis-pyrrolidine and bis-piperidine with the (S,S) absolute configuration as the major products (Scheme 3).
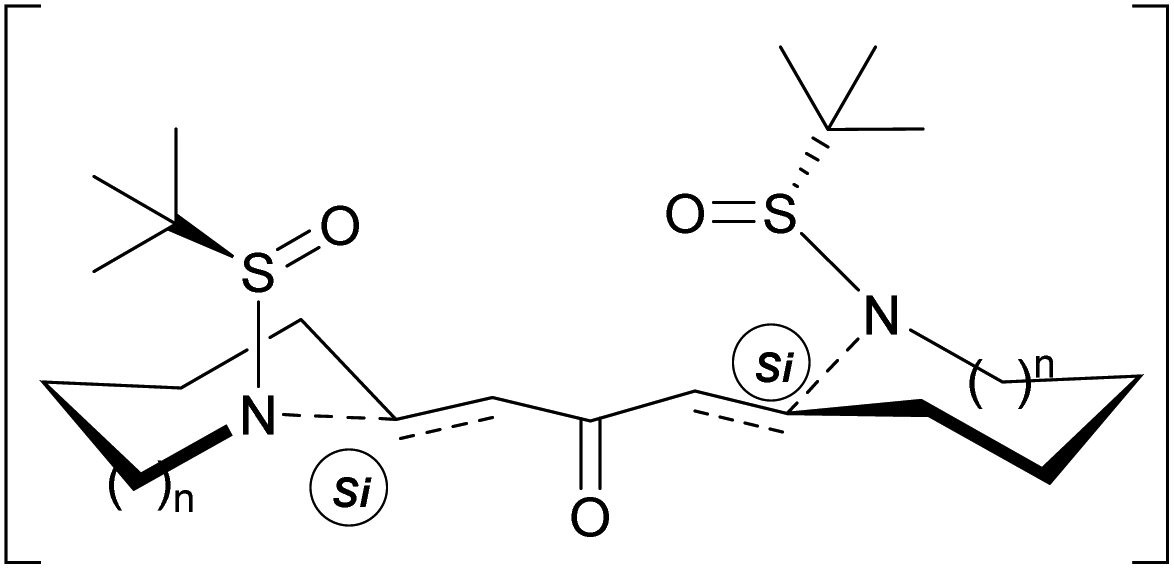 | ||
| Scheme 3 Stereochemical outcome. | ||
With bis-piperidine 8a in hand, removal of the sulfinyl moiety by treatment with HCl in dioxane rendered (+)-anaferine 1 in nearly quantitative yield (Scheme 4). Comparison of NMR data and optical rotation with those reported in the literature [found: [α]25D +48.7 (c 1.0 MeOH/H2O), lit.5 [α]22D −49.8 (c 1.0 MeOH/H2O)] led us to assign the S,S absolute configuration of the newly created stereocenters, which corresponds to the enantiomer of the natural product. It is important to point out that our strategy entails a significant improvement over the previous synthesis since the final product anaferine was obtained in three chemical steps and 70% overall yield from sulfinyl amine 3a (while the previous total synthesis of this alkaloid required 11 steps).
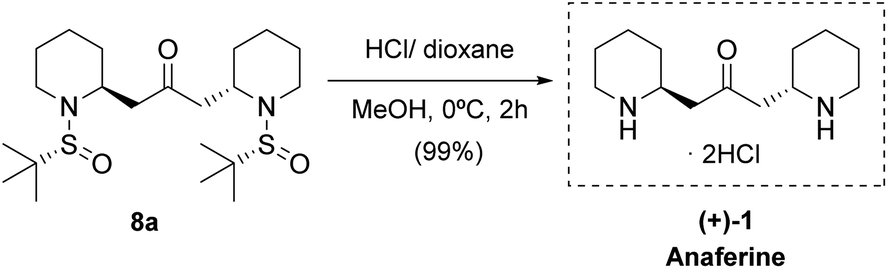 | ||
| Scheme 4 Synthesis of (+)-anaferine 1. | ||
On the other hand, bis-pyrrolidine 8b was transformed into (−)-dihydrocuscohygrine 2 in three chemical steps. Firstly, the carbonyl moiety was reduced with NaBH4 in methanol to afford compound 10 in 90% yield. Next, deprotection of the N atoms and further reductive amination with formaldehyde rendered (−)-dihydrocuscohygrine 2 in 86% yield (Scheme 5). NMR data and optical rotation were in good agreement with those previously reported in the literature [found: [α]25D −101.2 (c 1.5 acetone), lit.6 [α]22D −105.5 (c 1.67 acetone)]. In this way, the natural product dihydrocuscohygrine 2 was synthesized in five chemical steps and 45% overall yield from sulfinyl amine 3b.‡
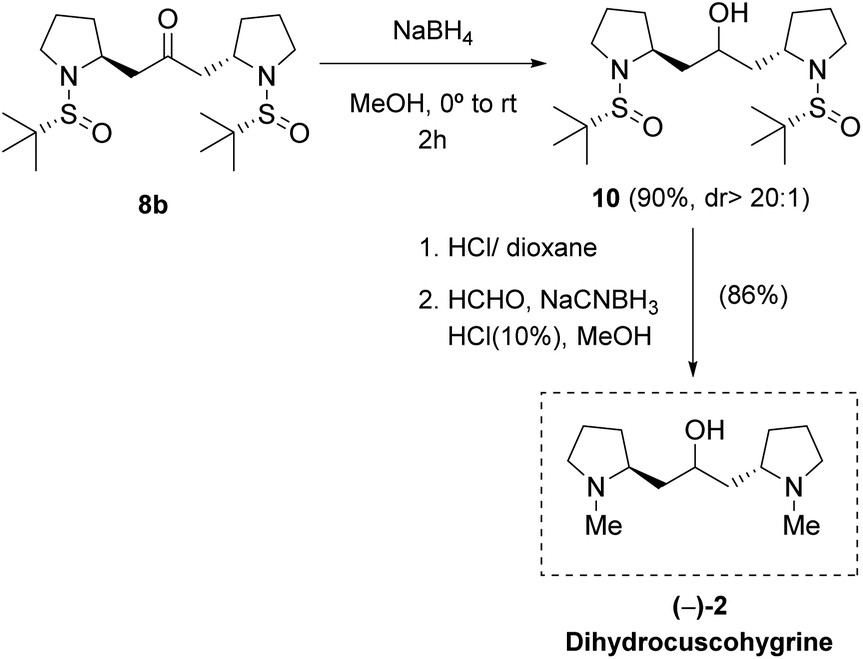 | ||
| Scheme 5 Synthesis of (−)-dihydrocuscohygrine 2. | ||
Conclusions
In summary, the total synthesis of alkaloids anaferine 1 and dihydrocuscohygrine 2 has been described employing a common approach. It takes advantage of a novel strategy that involves a bidirectional CM reaction followed by a double intramolecular aza-Michael reaction. The ability of N-sulfinyl amines to act as both nucleophilic nitrogen sources and chiral inducers was crucial for the stereoselective generation of the bis-piperidine and bis-pyrrolidine cores of these natural products. The synthesis described herein is the second one reported to date for anaferine and it involves three chemical steps in a highly convergent and efficient manner. Regarding dihydrocuscohygrine, it was synthesized in five chemical steps also in a very efficient manner.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the Spanish Ministerio de Economía y Competitividad (CTQ-2017-85026-R to C. P.). M. E., F. R.-A. and A. S.-V. thank the Spanish Ministerio de Educación, Cultura y Deporte for a predoctoral fellowships (FPU16/04533, FPU14/03520 and BES-2014-068186). J. T. thanks Generalitat Valenciana for a predoctoral fellowship (ACIF/2018/078).Notes and references
- (a) A. Rother, J. M. Bobbitt and A. E. Schwarting, Chem. Ind., 1962, 654 CAS; (b) A. E. Schwarting, J. M. Bobbitt, A. Rother, C. K. Atal, K. L. Khanna, J. D. Leary and W. G. Walter, Lloydia, 1963, 26, 258 CAS; (c) H. C. Beyerman, L. Maat and C. A. Moerman, Recl. Trav. Chim. Pays-Bas, 1971, 90, 1326 CrossRef CAS; (d) M. M. El-Olemy and A. E. Schwarting, J. Org. Chem., 1969, 34, 1352 CrossRef CAS.
- C. Remya, K. V. Dileep, E. J. Variayr and C. Sadasivan, Front. Life Sci., 2016, 9, 201 CrossRef CAS.
- G. Kumar and R. Patnaik, Med. Hypotheses, 2016, 92, 35 CrossRef CAS.
- (a) C. E. Turner, M. A. Elsohly, L. Hanus and H. N. Elsohly, Phytochemistry, 1981, 20, 1403 CrossRef CAS; (b) Y. M. A. El-Imam, W. C. Evans and T. Plowman, Phytochemistry, 1985, 24, 2285 CrossRef CAS; (c) P. Christen, M. F. Roberts, J. D. Phillipson and W. C. Evans, Phytochemistry, 1995, 38, 1053 CrossRef CAS.
- S. Blechert and C. Stapper, Eur. J. Org. Chem., 2002, 2855 CrossRef CAS.
- C. Stapper and S. Blechert, J. Org. Chem., 2002, 67, 6456 CrossRef CAS.
- T. Yamauchi, S. Hagiwara and K. Y. Higashiyama, J. Org. Chem., 2008, 73, 9784 CrossRef CAS PubMed.
- A. J. Wommack and J. S. Kingsbury, J. Org. Chem., 2013, 78, 10573 CrossRef CAS PubMed.
- For reviews highlighting the versatility of sulfinyl imines, see: (a) F. Foubelo and M. Yus, Chim. Oggi, 2016, 34, 45 CAS; (b) F. Foubelo and M. Yus, Eur. J. Org. Chem., 2014, 485 CrossRef CAS; (c) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS; (d) L. Jun and J. Hu, Future Med. Chem., 2009, 1, 875 CrossRef PubMed; (e) G.-Q. Lin, M.-H. Xu, Y.-W. Zhong and X.-W. Sun, Acc. Chem. Res., 2008, 41, 831 CrossRef CAS PubMed; (f) F. A. Davis, J. Org. Chem., 2006, 71, 8993 CrossRef CAS PubMed; (g) D. Morton and R. A. Stockman, Tetrahedron, 2006, 62, 8869 CrossRef CAS.
- For some representative examples, see: (a) F. A. Davis and J. Deng, Org. Lett., 2007, 9, 1707 CrossRef CAS PubMed; (b) F. A. Davis, Y. Wu, H. Yan, W. McCoull and K. R. Prasad, J. Org. Chem., 2003, 68, 2410 CrossRef CAS; (c) F. A. Davis, Y. Wu, H. Yan, K. R. Prasad and W. McCoull, Org. Lett., 2002, 4, 655 CrossRef CAS; (d) T. P. Tang and J. A. Ellman, J. Org. Chem., 2002, 67, 7819 CrossRef CAS PubMed. For a recent review, see: (e) J. R. Hernandez, F. Chemla, F. Ferreira, O. Jackowski, J. Oble, A. Pérez-Luna and G. Poli, Chimia, 2016, 70, 84 CrossRef CAS PubMed.
- S. Fustero, S. Monteagudo, M. Sánchez-Roselló, S. Flores, P. Barrio and C. del Pozo, Chem. – Eur. J., 2010, 16, 9835 CrossRef CAS PubMed.
- M. Guerola, M. Sánchez-Roselló, C. Mulet, C. del Pozo and S. Fustero, Org. Lett., 2015, 17, 960 CrossRef CAS PubMed.
- For reviews on the asymmetric aza-Michael reaction, see: (a) R. A. Yu, Russ. Chem. Bull., 2016, 65, 1687 CrossRef; (b) M. Sánchez-Roselló, J. L. Aceña, A. Simón-Fuentes and C. del Pozo, Chem. Soc. Rev., 2014, 43, 7430 RSC; (c) J. Wang, L. P. Li, P.-Y. Choi, A. S. C. Chan and F.-Y. Kwong, ChemCatChem, 2012, 4, 917 CrossRef CAS; (d) E. Reyes, M. Fernández, U. Uria, J. L. Vicario, D. Badia and L. Carrillo, Curr. Org. Chem., 2012, 16, 521 CrossRef CAS; (e) D. Enders, C. Wang and J. X. Liebich, Chem. – Eur. J., 2009, 15, 10058 CrossRef PubMed; (f) P. R. Krishna, A. Sreeshailam and R. Srinivas, Tetrahedron Lett., 2009, 65, 9657 CrossRef CAS.
- For the use of the aza-Michaael reaction in the synthesis of alkaloids, see: (a) Z. Amara, J. Caron and D. Joseph, Nat. Prod. Rep., 2013, 30, 1211 RSC; (b) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2019, 17, 3670 RSC.
- K. A. B. Austin, J. D. Elsworth, M. G. Banwell and A. C. Willis, Org. Biomol. Chem., 2010, 8, 751 RSC.
- For examples of DBU mediated aza-Michael reactions, see: (a) J. Yang, Y. Bao, H. Zhou, T. Li, N. Li and Z. Li, Synthesis, 2016, 48, 1139 CrossRef CAS; (b) F.-P. Huang, Z.-W. Dong, H.-R. Zhang and X.-P. Hui, J. Heterocycl. Chem., 2014, 51, 532 CrossRef CAS; (c) H. Quian, W. Zhao, H. H.-Y. Sung, I. D. Williams and J. Sun, Chem. Commun., 2013, 49, 4361 RSC; (d) J. Zhang, J. Wu, Z. Yin, H. Zeng, K. Khanna, C. Hu and S. Zheng, Org. Biomol. Chem., 2013, 11, 2939 RSC; (e) C.-E. Yeom, M. J. Kim and B. M. Kim, Tetrahedron, 2007, 63, 904 CrossRef CAS.
- For the previous use of TBAF as a base in aza-Michael-type additions, see: (a) S. Fustero, C. Báez, M. Sánchez-Roselló, A. Asensio, J. Miró and C. del Pozo, Synthesis, 2012, 44, 1863 CrossRef CAS; (b) S. Fustero, A. García-Sancho, G. Chiva, J. Sanz-Cervera, C. del Pozo and J. L. Aceña, J. Org. Chem., 2006, 71, 3299 CrossRef CAS PubMed; (c) G. V. M. Sharma, V. Goverdhan Reddy, S. Chander and K. Ravinder Reddy, Tetrahedron: Asymmetry, 2002, 13, 21 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo00811j |
| ‡ Our methodology to access dihydrocuscohygrine 2 is comparable in terms of number of steps and yield with those showed in Scheme 1, eqn (2) and (3). |
| This journal is © the Partner Organisations 2019 |