
Iron-mediated site-selective oxidative C–H/C–H cross-coupling of aryl radicals with quinones: synthesis of β-secretase-1 inhibitor B and related arylated quinones†
Adarsh Krishna
T.P
,
Sakthivel
Pandaram
and
Andivelu
Ilangovan
 *
*
Bharathidasan University, Tiruchirappalli, Tamilnadu-620024, India. E-mail: ilangovanbdu@yahoo.com
First published on 10th July 2019
Abstract
Phenoxy radicals generated from substituted arenes were converted into para site selective C-aryl radicals and coupled with quinones, using an inexpensive FeCl3–K2S2O8 system, to obtain several arylated quinones, in good to moderate yields, under operationally simple and mild conditions. This method is useful for one pot synthesis of β-secretase-1 (BACE) inhibitor B. The arylated quinones were used as intermediates in the synthesis of phenothiazin-5-ones. Theoretical studies on the pharmacokinetic properties of phenothiazin-5-ones showed high lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) p = 3.69), poor water solubility (logs = −6.42), and high gastrointestinal absorption (GI).
p = 3.69), poor water solubility (logs = −6.42), and high gastrointestinal absorption (GI).
Introduction
Quinone scaffolds gained importance owing to their roles in biological systems,1 and utilization as intermediates in biosynthesis,2 bioinspired synthetic products,1,3 and materials.4 Arylated quinones, especially phenol derivatives (Fig. 1), show inhibition of Alzheimer's disease (AD), through halting the production of β-amyloid peptides generated by β-secretase (BACE),5a and other interesting biological activities.1,5 | ||
| Fig. 1 Selected biologically active quinone derivatives. | ||
Owing to their importance, several approaches, involving different types of starting materials, were described for the synthesis of C-arylated quinones. Utilisation of pre-functionalised quinones and pre-functionalised arenes requires the presence of expensive transition metal reagents. Examples include Pd2(dba)3-CuI-Ph3As6a catalyzed coupling of a stannylquinone with an arylhalide, Pd(PPh3)4-CuI5b/Pd(PPh3)4-CuBr6b catalyzed coupling of a haloquinone with a styrylstannane, and Pd(dppf)Cl26c/Pd(PPh3)46d,e/Pd(OAc)26f catalyzed Suzuki coupling.
Heck type arylation often comprises unfunctionalized quinones and pre-functionalized arenes. Commercially available, stable, but costly arylboronic acids (Fig. 2) were used in such transformations, in the presence of expensive reagents made of metals such as Ag,7a Rh,7b Ir,7c and Pd,7d,e and also inexpensive Fe salts.7f–h Furthermore, in addition to metal free arylation of quinones, using arylboronic acid-K2S2O8, developed by us,8a several metal free methods using diazonium salt-hydrazine catalysts,8b diazonium salts,8c,d diaryliodonium salts,8e and arylhydrazine salts8f have also been described. Recently, metal mediated C–H arylation of quinones using triazenes (Ag),9a aryl halides (Pd),9b and aniline (CuO-NPs/Gr9c or PANI-g-C3N4-TiO2)9d has been demonstrated.
 | ||
| Fig. 2 Methods of synthesis of arylated naphthoquinones. | ||
On the other hand, oxidative C–H/C–H coupling, a fast developing and straightforward concept, used in assembling a diverse variety of complex aromatic systems,10a was also employed in coupling of quinones and arenes. Such C–H/C–H coupling was yet again carried out in the presence of costly metal salts such as highly electrophilic Pd(OAc)2,10b,c Rh salts,10d and Pd(TFA)2.10e Conversely, ligand like binding of quinones with metal catalysts, the redox nature, the need for use of expensive reagents, and lengthy steps involved made such procedures disadvantageous.
In essence, this literature background shows that arene–quinone coupling was demonstrated using either organometallic reagents generated from prefunctionalised/unfunctionalised starting materials and expensive transition metals or aryl radicals obtained from prefunctionalised arenes. Consequently, there is no report known on the use of aryl radicals generated through oxidative C–H bond cleavage. This impelled us to make use of aryl radicals generated via oxidative C–H cleavage of electron rich arenes.
Aromatic compounds containing multiple C–H bonds make site-selective arylation a challenging task. Over the past few years meta11 or ortho12 or para13 site selective C–H activation of aromatics has been successfully demonstrated using steric, electronic, catalytic and directing group effects. Phenol directed C–H functionalization reactions involve a number of different mechanistic forms.14 Oxidative cross-coupling of phenols often leads to the formation of homocoupling by-products, higher-molecular weight polymers, and C,O-connected phenols, depending upon the oxidant and the experimental conditions. Thus controlling such reactions remains a challenge.12b Recently, ortho C–H annulation of 4-methoxy phenol radicals (generated via oxidative C–H cleavage) with olefins has been described.12b However, when we examined the coupling of 4-OMe-Ar-OH (2r) with 1,4-naphthoquinone (1a) in the presence of different concentrations of a FeCl3 (0.1 or 1 or 3 equiv.) and DDQ (1.2 equiv.) system,12b no product was obtained. Furthermore, in deviation from the literature methods, and in continuation of our interest in developing new methods for arylation of quinones,8a,b we have identified para site selective coupling of an arene with a quinone in the presence of an inexpensive FeCl3–K2S2O8 system. Herein, we disclose the details.
Results and discussion
Since a para substituted phenol (4-OMe-Ar-OH (2r)) failed to elicit the desired product, alternatively, coupling of a mono-substituted phenol (2a, 1.0 mmol.) with napthoquinone (1a, 0.5 mmol.) was examined employing the FeCl3 (0.1 equiv.)–DDQ (1.2 equiv.) system.12b This reaction at rt lead to no product (Table 1, entry 1). Interestingly, increasing FeCl3 to a stoichiometric amount (1.0 equiv.) led to the formation of product 3a, para site selectively, only in 30% yield. Given that, it would be an immoderate approach to use a quinone derivative (DDQ) in more than a stoichiometric quantity for arylation of another quinone, and the product is obtained only in low yield, we examined alternative oxidants. Notably, when a cheap and readily available oxidant K2S2O8 was employed, at rt, in incremental concentrations (1.0/2.0/3.0 equiv., entry 2) with FeCl3 (1.0 equiv.) product 3a was obtained in 39%, 51% and 60% yields, respectively. However, when the quantity of the mediator (FeCl3) was increased to 2.0 (entry 3) and 3.0 equiv. (entry 4) the yield of the product 3a increased substantially and the highest yield (86%) was obtained when 3.0 equiv. of FeCl3 and 3 equiv. of K2S2O8 were used. Furthermore, when the temperature was raised to 60° C the yield decreased to 82% (entry 4), indicating that higher temperature would be detrimental to the reaction.|
|
||||
|---|---|---|---|---|
| Entry | Mediator | Oxidant/additive | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol); 2a (1.0 mmol); solvent (4 mL); mediator and/or oxidant time 12 h, rt. b Isolated yield; NR = no reaction. c Quantity of the mediator or oxidant: 0.1 equiv. d Quantity of the mediator or oxidant: 1.2 equiv. e Quantity of the mediator or oxidant: 1.0 equiv. f Quantity of the mediator or oxidant: 2.0 equiv. g Quantity of the mediator or oxidant: 3.0 equiv. h Quantity of the mediator or oxidant: 4.0 equiv. i Quantity of the mediator or oxidant: 5.0 equiv. j Quantity of the mediator or oxidant: 0.5 equiv. k Temperature – 60 °C; oxidant: 3.0 equiv. l X = Zn, Sn, Cu and Ni. | ||||
| 1 | FeCl3c,e | DDQd | Tol | NRc/30e |
| 2 | FeCl3e | K2S2O8e,f,g | DCM | 39e/51f/60g |
| 3 | FeCl3f | K2S2O8e,f,g | DCM | 56 e/64f/68g |
| 4 | FeCl3g | K2S2O8e,f,g | DCM | 68e/72f/86g/82k |
| 5 | FeCl3h,i | K2S2O8g | DCM | 82h/55i |
| 6 | FeCl3g | (NH4)2S2O8e | DCM | 44f |
| 7 | FeCl3g | Oxonee | DCM | 62f |
| 8 | FeCl3g | H2O2e | DCM | 20f |
| 9 | FeCl3g | TBHPe | DCM | 40f |
| 10 | FeCl3g | K2S2O8e | ACN/THF | Trace |
| 11 | FeCl3g | K2S2O8e | MeOH/H2O | NR |
| 12 | FeCl3e,g/FeSO4e | — | DCM | 33e/57g/Trace |
| 13 | XCl2l,e/AlCl3e | — | DCM | NR |
| 14 | TFAg/MsOHg/pTSAg | — | DCM | NR/Trace/15 |
| 15 | FeCl3e | pTSAg | DCM | 35 |
| 16 | — | K2S2O8g | DCM | Mixture |

With an additional increase in the quantity of FeCl3 (4.0 equiv. and 5.0 equiv., entry 5) the yield of product 3a decreased to 82% and 55%, respectively. Furthermore, reactions carried out with 3.0 equiv. FeCl3 and 1.0 equiv. of various oxidants such as (NH4)2S2O8, oxone, H2O2 and TBHP (entries 5–8), did not lead to an improvement in yield compared to that with K2S2O8. Subsequently, examination of different solvents and their combination revealed that DCM is the best solvent (entries 10 and 11) to provide good yield.
Furthermore, we examined the possibility of Friedel–Crafts type arylation using different types of acids. When Lewis acid FeCl3 (1.0 equiv. or 3.0 equiv., entry 12) alone was used, the desired product 3a was obtained in 33% and 57% yields, respectively. Other Lewis acid promoters such as FeSO4, ZnCl2, SnCl2, CuCl2·2H2O, NiCl2·6H2O and AlCl3 were found to be ineffective (entries 12 and 13). Among the Brønsted acids only PTSA elicited product 3a albeit in low yield (entry 15, 15%). A combination of Lewis acid FeCl3 and Brønsted acid PTSA provided a low yield of the product 3a (35%). No desired product 3a was found when K2S2O8 (3.0 equiv.) alone was employed. Since product 3a was obtained, via the Friedel–Crafts pathway, only in moderate yield (57%) using FeCl3 (3.0 equiv.) as the mediator, this approach was abandoned. Finally, FeCl3 (3.0 equiv.) and K2S2O8 (3.0 equiv.) in DCM, which helps in generation of the aryl radical at rt, was identified as the optimal reaction condition for further study. FeCl3 is expected to help in conversion of the phenoxy radical generated into a C radical and help moderate the reaction.
Interestingly, compound 3a, an effective β-secretase-1 inhibitor B (IC50 = 8.22 μm),5a has been reported to be synthesised in two steps involving deprotection using BBr3 in moderate overall yield.7c,9b,e However, our synthetic approach provides the same compound 3a in high yield (86%), in one pot, and is found to be simple and efficient. Next, we examined the scope and limitations of the FeCl3 mediated C–H arylation of quinones using electron-rich arenes (phenol/anisole, Table 2). While o-cresol provided the desired product 3b in high yield (89%), sterically hindered m-cresol provided product 3c in moderate yield (47%). 2,5-Dimethyl phenol (2d) underwent the reaction smoothly to afford the product 3d in moderate yield (43%). Electron rich but sterically less hindered compounds 2-allylphenol (2e) and 2,6-di-tert-butylphenol (2f) provided the corresponding products 3e (82%) and 3f (92%) in excellent yields. Highly reactive catechol (2g) and 3-methylcatechol (2h) provided products 3g and 3h in low (34%) and moderate (60%) yields. Unremarkably, phenols 2i (2-NO2), 2j (2-CHO), 2k (2-COMe), 2l (2-Cl), 2m (2-Br), and 2n (2-I) substituted with either strong or moderately deactivating substituents provided no corresponding products 3i–n even after 24 h.
| a Reaction conditions: 1 (1.0 equiv.), 2 (2.0 equiv.), FeCl3 (3.0 equiv.), K2S2O8 (3.0 equiv.), DCM (4 mL), rt, time: 6–24 h. Isolated yield. Gram scale 1a (1.0 g). |
|---|

|
Interestingly, the methylether of phenol 2o, underwent reaction with 1a to afford product 3o in good (77%) yield. Nevertheless, 2-bromo-naphthoquinone (1b), containing inductively electron withdrawing substituent 2-Br, offered product 3p in low yield (38%), after 24 h. However, naphthoquinone 1c–e containing 2-OH, 2-NH2, and 2-CH3 substituents underwent no reaction with anisole (2o), which indicates that electron donating substituents at a conjugating position to the reactive site do not favour the coupling reaction. Furthermore, on treating 1a separately with dimethoxy benzene (2p) and trimethoxy benzene (2q), the desired products 3t and 3u, respectively, were obtained in good yields. Subsequently, when the reaction of anisole (2i) was examined on 1,4-benzoquinone a mixture of products was obtained. However, the reaction of anisole (2i) with mono- or di-substituted 1,4-benzoquinone such as tert-butyl 1f and 1g afforded the desired products 3v (43%) and 3w (35%), respectively, in moderate yields. The reaction of 1a with benzene, as well as alkyl arenes such as xylene or toluene, provided no product. These results shows that the presence of an –OH or –OMe group is important for this oxidative coupling reaction. Moreover, gram-scale experiments carried out between 1a (1.0 g) and 2a or 2o provided products 3a and 3o in 70% and 66%, respectively. This shows that the present method can be easily adopted for large scale preparation.
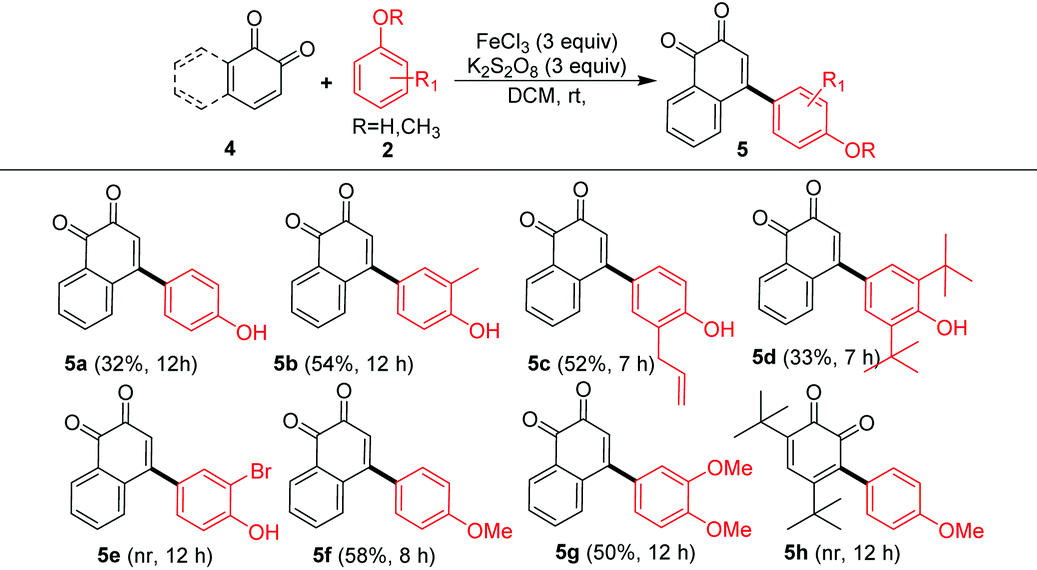
Having examined the reaction of 1,4-quinones, next, the scope of the arylation was studied on 1,2-napthoquinone (4a) and the results are summarized in Table 3. In this case also we observed C–C bond formation but not C–O bond formation as reported by Lumb et al.15 Products corresponding to phenol (5a, 32%), o-cresol (5b, 54%), allyl phenol (5c, 52%) and 2,6-di-tert-butylphenol (5d, 33%) were obtained in moderate yields. However, similar to our previous observation (Scheme 2, entry 3q), 2-bromophenol (2m) failed to deliver the desired product 5e. Furthermore, anisole (2o) and veratrole (2p) underwent the reaction to deliver desired products 5f (58%) and 5g (50%) in good yields. In another reaction, highly sterically hindered 3,5-di-tert-butyl-1,2-benzoquinone (4d) unsuccessfully reacted with anisole (2o) and the desired product 5h could not be obtained.
| a Reaction conditions: 4 (1.0 equiv.), 2 (2.0 equiv.), FeCl3 (3.0 equiv.), K2S2O8 (3.0 equiv.), DCM (4 mL), RT, time 7–12 h. Isolated yield. |
|---|
|
The tert-butyl group plays an important role in chemistry and biology.16 Interestingly, when TBHP (1.0 equiv.) was used as an oxidant instead of K2S2O8, two reactions, namely, tert-butyl group insertion at the C-2 position and oxidative C–H/C–H cross-coupling, took place to form unexpected product 3x (34%) along with the expected product 3a (40%). Furthermore, when tert-butyl hydroquinone (6a) was treated with 2o, a chain of oxidation reactions, namely, hydroquinone to quinone conversion and oxidative C–H/C–H cross-coupling took place in one pot to form product 3v in moderate yield (Scheme 1).
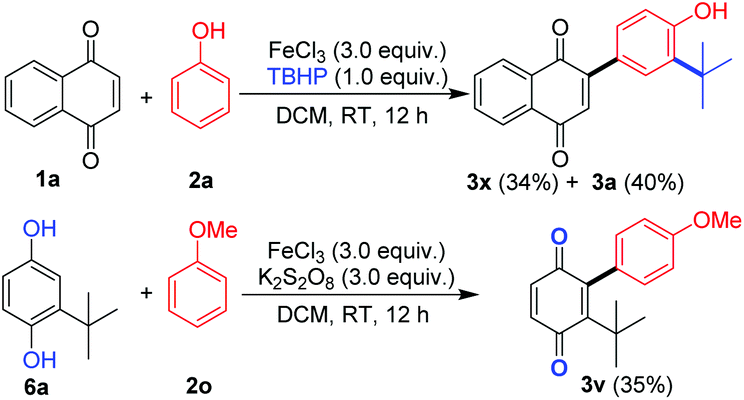 | ||
| Scheme 1 Synthesis of aryl quinones with a tert-butyl group. | ||
Additionally, the product 3p was further used as a starting material for the synthesis of a new phenothiazin-5-one derivative (Scheme 2).17a Accordingly, compound 3p was treated with 2-amino-4-chlorobenzenethiol at 100 °C in the presence of a base to obtain the corresponding phenothiazin-5-one 8a in excellent yield.
 | ||
| Scheme 2 Synthesis of a phenothiazin-5-one derivative. | ||
Furthermore, the pharmacokinetic properties of 8a, via absorption, distribution, metabolism and excretion (ADME), were predicted using DataWarrior software (Version 4.7.2),17b and the SwissADME web tool17c (see the ESI†). An in silico study revealed that 8a showed high lipophilicity (logp = 3.69), poor water solubility (logs = −6.42), and high gastrointestinal absorption (GI). These results show that compound 8a, falling in Class II described by Amidon et al.,17d does not have blood brain barrier (BBB) permeability and has less skin permeation (logKp = −4.73 cm s−1). Compound 8a was found to show inhibition of four cytochrome P450 isomers (CYP1A2, CYP2C19, CYP2C9 and CYP3A4), exhibit a good bioavailability value (0.55) and meet the criteria of drug-likeness assessment based on Lipinski and Veber rules. Further in vitro studies of this compound are in progress in our laboratory.
Next, to gain understanding about the mechanism, control experiments were carried out (Scheme 3). In the absence of FeCl3 and only with K2S2O8 the reaction between compounds 1a and 2a failed (equiv. 1). As shown earlier (Table 1, entries 12 and 15) in the presence of FeCl3 alone or FeCl3 and pTSA product 3a is formed, and it must have been formed through the Friedel Crafts arylation mechanism. Furthermore, when 2.0 equiv. of a radical trapping agent such as TEMPO were added, the reaction between compounds 1a and 2a or 2o failed to deliver the desired product 3a or 3o (equiv. 2), which clearly indicated that, in the presence of an excess of the oxidant (K2S2O8), the reaction takes place via the radical pathway and not through Friedel–Crafts arylation (Scheme 4, path C).
 | ||
| Scheme 3 Verification experiments for the mechanism. | ||
 | ||
| Scheme 4 Plausible mechanism. | ||
When the para position of the phenol was blocked as in the case of p-methyl phenol (2r) and p-methoxy phenol (2s) (equiv. 3), no ortho C–H functionalised products 3y and 3z or any other product was obtained, indicating that this reaction is para selective. All the forgoing discussion indicates that this FeCl3 and K2S2O8 mediated oxidative C–H/C–H coupling reaction is a radical process and para site-selective in C–H functionalization of phenol.
Based on these results and previous literature background, a plausible reaction mechanism is proposed (Scheme 4). Initially, in the presence of FeCl3, K2S2O8 dissociates to generate a highly reactive sulfate radical (SO4˙−K+),7f–h which in turn reacts with phenol (2a) to form a phenoxy radical (A). Furthermore, FeCl3 promotes the conversion of the O-radical to C-radical (B)12b and helps its stabilization through resonance. The C-radical (B) on reaction with 1a gives rise to intermediate (C), which reoxidises to attain a more stable quinone form and afford arylated product 3a (Scheme 4, path A). In a similar manner, –OMe substituted aromatics are expected to undergo reaction through the radical cation mechanism (Scheme 4, path B).10a
Conclusion
An efficient method for para site-selective C–H/C–H coupling of electron rich arenes with quinones under mild conditions is demonstrated. A series of arylated quinones were obtained in moderate to excellent yields at rt and they are also scalable to the gram scale level. To the best of our knowledge this is the first example in which an aryl radical generated through C–H cleavage is used in arylation of quinones. FeCl3 promotes the O-radical to C-radical conversion and C-arylation reaction. This eco-friendly, inexpensive FeCl3–K2S2O8 mediator system tolerates a range of functional groups. Furthermore, β-secretase-1 inhibitor-B and related analogs were obtained in one pot. Arylated quinones were used as starting materials for the synthesis of phenothiazin-5-one derivatives which meets the requirement of drug-likeness which was studied using DataWarrior software and the SwissADME web tool. Further study on making use of this protocol is in progress in our research.Experimental
General information
All the reagents were purchased commercially and used without further purification. 1H NMR (400 MHz) and 13C NMR (100 MHz) were recorded with a Bruker 400 MHz spectrometer in CDCl3 with tetramethylsilane (TMS) as the internal standard. Multiplicities are reported using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, sep = septet, br = broad resonance. All the NMR spectra were acquired at ambient temperature. Analytical thin layer chromatography (TLC) was performed using Silica Gel 60 Å F254 pre-coated plates (0.25 mm thickness). Visualization was accomplished by irradiation with a UV lamp and staining with I2 on silica gel. High resolution mass spectra (HRMS) were recorded on a Thermo Executive Plus spectrometer.2-(4-Hydroxyphenyl)naphthalene-1,4-dione (3a). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (orange solid, 136 mg, 86.0% yield, mp 172–174 °C); 1H NMR (400 MHz, CDCl3): δ 9.00 (s, 1H), 8.13–8.11 (m, 1H), 8.06–8.04 (m, 1H), 7.73–7.70 (m, 2H), 7.46 (d, J = 8.0, 2H), 6.98 (s, 1H), 6.90 (d, J = 8.0, 2H); 13C NMR (100 MHz, CDCl3): δ 185.3, 185.0, 159.5, 147.7, 133.70, 133.68, 133.1, 132.7, 132.1, 131.2, 127.0, 125.8, 124.4, 115.8. HRMS (ESI): m/z [M + H]+ calcd for C16H10O3: 250.0630; found: 250.0631.
2-(4-Hydroxy-3-methylphenyl)naphthalene-1,4-dione (3b). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 148.33 mg, 89.0% yield, mp 170–172 °C); 1H NMR (400 MHz, CDCl3 and one drop of DMSO-D6): δ 9.00 (s, 1H), 8.02–8.00 (m, 2H), 7.64–7.62 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 6.90 (s, 1H), 6.81 (d, J = 8.0 Hz, 1H), 2.16 (s, 3H); 13C NMR (100 MHz, CDCl3 and one drop of DMSO-D6): δ = 185.1, 185.0, 157.8, 147.7, 133.6, 133.5, 132.8, 132.6, 132.1, 132.0, 128.5, 126.8, 125.6, 124.9, 124.1, 114.9, 16.2 HRMS (ESI): m/z [M + H]+ calcd for C17H12O3: 264.0786; found: 264.0776.
2-(4-Hydroxy-2-methylphenyl)naphthalene-1,4-dione (3c). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10, (78.64 mg, 47.0% yield, mp 130–132 °C); 1H NMR (400 MHz, DMSO-D6): δ = 8.00–7.95 (m, 2H), 7.85–7.82 (t, 2H), 7.01 (d, J = 8.0 Hz, 2H), 6.82 (s, 1H), 6.68–6.63 (m, 2H), 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 185.5, 184.6, 156.9, 150.4, 138.3, 136.8, 134.0, 132.3, 132.1, 130.9, 127.1, 126.2, 125.9, 117.5, 112.9, 20.7; HRMS (ESI): m/z [M + H]+ calcd for C17H12O3: 264.0786; found: 264.0776.
2-(4-Hydroxy-2,5-dimethylphenyl)naphthalene-1,4-dione (3d). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 75 mg, 43.0% yield, mp 126–127 °C); 1H NMR (400 MHz, CDCl3): δ 8.16–8.12 (m, 2H), 7.79–7.77 (m, 2H), 6.93 (d, J = 12 Hz, 2H), 6.66 (s, 1H), 5.88 (s, 1H), 2.21 (s, 3H), 2.13 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 185.5, 184.8, 155.1, 150.4, 136.7, 135.4, 133.9, 132.3, 132.1, 127.1, 126.1, 125.7, 121.5, 117.1, 20.2, 15.3, HRMS (ESI): m/z [M − H]− calcd for C18H14O3: 278.0943; found: 278.0944.
2-(3-Allyl-4-hydroxyphenyl)naphthalene-1,4-dione (3e). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (orange solid, 150.55 mg, 82.0% yield, mp 164–165 °C); 1H NMR (400 MHz, DMSO-D6): δ 8.00–7.81 (m, 2H), 7.74–7.72 (m, 2H), 7.22 (d, J = 8.0, 2H), 6.82–6.79 (t, 2H), 5.94–5.83 (s, 1H), 4.97–4.95 (m, 2H), 3.22 (d, J = 6.0, 2H); 13C NMR (100 MHz, CDCl3): δ 185.3, 185.0, 159.5, 147.7, 133.7, 133.6, 133.1, 132.6, 132.1, 131.2, 126.9, 125.8, 124.4, 115.8. HRMS (ESI): m/z [M − H]− calcd for C19H14O3: 290.0943; found: 290.0946.
2-(3,5-Di-tert-butyl-4-hydroxyphenyl)naphthalene-1,4-dione (3f). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (orange red solid, 210.9 mg 92% yield, mp 152–151 °C); 1H NMR (400 MHz, CDCl3): δ = 8.16–8.14 (m, 1H), 8.08–8.06 (m, 1H), 7.72–7.70 (m, 2H), 7.47 (s, 2H), 7.04 (s, 1H), 5.62 (d, 1H), 1.5 (s, 18H). 13C NMR (100 MHz,): δ = 185.3, 185.1, 156.1, 148.5, 136.0, 133.7, 133.4, 132.7, 132.2, 127.1, 126.9, 125.9, 124.6, 34.5, 30.3. HRMS (ESI): m/z [M − H]− calcd for C24H26O3: 362.1882; found: 362.1884.
2-(3,4-Dihydroxyphenyl)naphthalene-1,4-dione (3g). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (brown solid, 57.25 mg, 34.0% yield, mp 240–241 °C); 1H NMR (400 MHz, DMSO-D6): δ = 8.08–8.06 (m, 1H), 8.01–7.99 (m, 1H), 7.90–7.87 (m, 2H), 7.11 (d, J = 4.0 Hz 1H), 7.02–7.0 (m, 2H), 6.85 (d, J = 8.0 Hz, 1H). 13C NMR (100 MHz,): δ = 184.5, 184.4, 147.6, 147.0, 144.8, 134.0, 132.4, 132.3, 131.5, 126.5, 125.2, 124.2, 121.6, 117.0, 115.4. HRMS (ESI): m/z [M + H]+ calcd for C16H10O4: 266.0579; found: 266.0581.
2-(3,4-Dihydroxy-2-methylphenyl)naphthalene-1,4-dione (3h). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 106.34 mg, 60.0% yield, mp 182–184 °C); 1H NMR (400 MHz, DMSO-D6): δ = 8.08–8.06 (m, 1H), 8.00–7.98 (m, 1H), 7.90–7.87 (m, 2H), 6.99–6.98 (t, 2H), 6.93 (s, 1H), 2.16 (s, 3H). 13C NMR (100 MHz): δ = 189.8, 189.8, 152.4, 151.1, 149.5, 139.2, 137.6, 136.8, 131.7, 130.5, 129.6, 128.6, 128.3, 119.7, 21.2. HRMS (ESI): m/z [M + H]+ calcd for C17H12O3: 280.0736; found: 280.0739.
2-(4-Methoxyphenyl)naphthalene-1,4-dione (3o). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (orange yellow solid, 128.80 mg, 77.08% yield, mp 134–135 °C); the product was obtained as an orange yellow solid after being subjected to short silica gel column chromatography (hexane/ethyl acetate, 90:10). 1H NMR (400 MHz, CDCl3): δ 8.16–8.14 (m, 2H), 8.10–8.07 (m, 2H), 7.80–7.74 (m, 2H), 7.57 (d, J = 8.0 Hz, 2H), 7.27 (s, 1H), 7.00 (d, J = 8.0 Hz, 2H), 3.85 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 185.2, 184.8, 161.3, 147.4, 133.8, 133.7, 132.6, 132.1, 131.1, 127.0, 125.9, 125.7, 114.0, 55.4; HRMS (ESI): m/z [M + H]+ calcd for C17H12O3: 264.0786; found: 264.0406.
2-Bromo-3-(4-methoxyphenyl)naphthalene-1,4-dione (3p). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (orange solid, 55.30 mg, 38.0% yield, mp 126–127 °C); 1H NMR (400 MHz, CDCl3): δ 8.22–8.2 (m, 1H), 8.15–8.13 (m, 1H), 7.79–7.77 (m, 2H), 7.33 (d, J = 8.0 Hz, 2H), 7.01 (d, J = 8.0 Hz, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 181.8, 178.4, 160.5, 149.3, 138.5, 134.4, 134.1, 131.7, 131.6, 131.3, 131.2, 127.5, 127.4, 126.0, 113.5, 55.4.
2-(3,4-Dimethoxyphenyl)naphthalene-1,4-dione (3t). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 130.30 mg, 70.0% yield, mp 132–134 °C); 1H NMR (400 MHz, CDCl3): δ = 8.14–8.12(m, 1H), 8.08–8.05 (m, 1H), 7.75–7.72 (m, Hz, 2H), 7.22–7.20 (dd, J1 = 4.0, J2 = 4.0 Hz, 1H), 7.14–7.13 (d, J = 4.0 Hz 1H), 7.02 (s, 1H), 6.94–6.92 (d, J = 8.0 Hz, 1H), 3.91–3.91(s, 6H); 13C NMR (100 MHz, CDCl3): δ = 185.1, 184.7, 150.9, 148.7, 147.3, 133.9, 133.8, 138.1, 127.0, 125.9, 125.8, 122.9, 112.4, 110.9, 56.0, 56.0; HRMS (ESI): m/z [M + H]+ calcd for C18H14O3: 294.0892; found: 294.0898.
2-(2,3,4-Trimethoxyphenyl)naphthalene-1,4-dione (3u). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (yellow solid, 78.64 mg, 47.0% yield, mp 85–87 °C); 1H NMR (400 MHz, CDCl3): δ = 8.16–8.09 (m, 2H), 7.76–7.74 (m, 2H), 6.99 (s, 1H), 6.97 (d, J = 4.0 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.86 (s, 3H). 13C NMR (100 MHz,): δ = 185.1, 184.0, 155.4, 151.9, 147.9, 142.0, 132.6, 133.8, 133.6, 132.6, 132.2, 126.9, 126.0, 124.9, 120.9, 107.0, 61.1, 60.8, 56.1.
6-(tert-Butyl)-4′-methoxy-[1,1′-biphenyl]-2,5-dione (3v). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (orange yellow solid, 70.82 mg, 43.0% yield, mp 242–243 °C); 1H NMR (400 MHz, CDCl3): δ = 7.64 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 8.0 Hz, 2H), 6.66 (d, J = 4.0 Hz, 1H), 6.65 (d, J = 4.0 Hz 1H), 3.76 (s, 3H), 1.25 (s, 9H). 13C NMR (100 MHz,): δ = 188.4, 187.2, 161.1, 156.3, 147.3, 131.5, 130.5, 130.0, 128.8, 125.9, 113.9, 55.4, 35.6, 29.4. HRMS (ESI): m/z [M + H]+ calcd for C17H18O3: 270.1256; found: 270.1258.
3,6-Dichloro-4′-methoxy-[1,1′-biphenyl]-2,5-dione (3w). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (orange red solid, 56.11 mg, 35.0% yield mp 135–136 °C); 1H NMR (400 MHz, CDCl3): δ = 7.28 (d, J = 8.0 Hz, 2H), 7.21 (s, 1H), 7.00 (d, J = 8.0 Hz, 2H), 3.86 (s, 3H). 13C NMR (100 MHz,): δ = 177.8, 177.4, 160.9, 144.4, 143.2, 140.0, 133.0, 131.7, 122.7, 113.7, 55.4.
2-(3-(tert-Butyl)-4-hydroxyphenyl)naphthalene-1,4-dione (3x). The reaction was carried out according to general method B. Eluent, hexane/ethyl acetate = 90
:10 (orange red semisolid, 65 mg, 34.0% yield); 1H NMR (400 MHz, CDCl3): δ = 8.19–8.17 (m, 1H), 8.12–8.10 (m, 1H), 7.78–7.75 (m, 2H), 7.53 (d, J = 4.0 Hz, 1H), 7.38–7.35 (m, 1H), 7.05 (s, 1H), 6.79–6.77 (d, J = 8.0 Hz, 1H), 5.68 (s, 1H), 1.5 (s, 9H); 13C NMR (100 MHz, CDCl3): δ = 185.5, 185.1, 156.6, 148.1, 136.5, 133.8, 133.5, 132.7, 132.2, 128.9, 128.7, 127.0, 125.9, 125.4, 116.7, 34.7, 29.5; HRMS (ESI): m/z [M + H]+ calcd for C16H10O3: 306.1256; found: 306.1250.
4-(4-Hydroxyphenyl)naphthalene-1,2-dione (5a). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 25.31 mg, 32.0% yield, mp 167–168 °C); 1H NMR (400 MHz, DMSO-D6): δ = 7.97 (d, J = 7.2 Hz, 1H), 7.66–7.53 (m, 2H), 7.31 (s, 1H), 7.30 (d, J = 8.0 Hz 2H), 6.90 (d, J = 8.0 Hz, 2H), 6.22 (s, 1H); 13C NMR (100 MHz, CDCl3 and one drop of DMSO-D6): δ = 180.4, 179.9, 159.4, 157.5, 135.2, 135.0, 131.7, 130.7, 130.3, 129.9, 129.8, 126.6, 115.9; HRMS (ESI): m/z [M + H]+ calcd for C16H10O3: 250.0630; found: 250.0629.
4-(4-Hydroxy-3-methylphenyl)naphthalene-1,2-dione (5b). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 45.11 mg, 54.0% yield mp 192–193 °C); 1H NMR (400 MHz, DMSO-D6): δ = 7.99 (d, J = 8.0 Hz, 1H), 7.67–7.55 (m, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.19 (s, 1H), 7.15–7.13 (m, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.22 (s, 1H), 2.14 (s, 3H); 13C NMR (100 MHz, CDCl3 with one drop of DMSO-D6): δ = 180.5, 180.1, 157.7, 157.4, 135.4, 135.0, 131.7, 130.9, 130.6, 130.3, 129.9, 127.3, 127.2, 126.6, 125.3, 115.1, 16.2.
4-(3-Allyl-4-hydroxyphenyl)naphthalene-1,2-dione (5c). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 48 mg, 52.0% yield, mp-); 1H NMR (400 MHz, DMSO-D6): δ = 7.99 (d, J = 8.0 Hz, 1H), 7.66–7.62 (m, 1H), 7.57–7.53 (m, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.18–7.14 (m, 2H), 6.92 (d, J = 8.0 Hz, 1H), 6.20 (s, 1H), 5.96–5.89 (m, 1H), 5.03–4.98 (m, 2H), 3.29 (d, 2H); 13C NMR (100 MHz, CDCl3 and one drop of DMSO-D6): δ = 180.4, 180.1, 157.7, 157.1, 136.3, 135.4, 135.0, 131.7, 130.6, 130.3, 130.1, 129.8, 127.6, 127.3, 126.6, 115.9, 115.4, 96.1, 34.1; HRMS (ESI): m/z [M + H]+ calcd for C16H10O3: 290.0943; found: 290.0947.
4-(3,5-Di-tert-butyl-4-hydroxyphenyl)naphthalene-1,2-dione (5d). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (reddish brown solid, 38.0 mg 33.0% yield, mp 214–215 °C); 1H NMR (400 MHz, CDCl3): δ = 8.22–8.20 (m, 1H), 7.63–7.60 (m, 1H), 7.56–7.52 (m, 1H), 7.45 (d, J = 8.0 Hz, 1H), 6.45 (s, 1H), 5.56 (s, 1H), 1.5 (s, 18H). 13C NMR (100 MHz,): δ = 180.6, 180.1, 158.2, 155.6, 136.4, 135.5, 135.0, 132.0, 130.6, 129.8, 127.6, 127.0, 125.4, 34.5, 30.3. HRMS (ESI): m/z [M − H]− calcd for C24H26O3: 362.1882; found: 362.1887.
4-(4-Methoxyphenyl)naphthalene-1,2-dione (5f). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (yellow solid, 48.45 mg, 58.0% yield, mp 128–129 °C); 1H NMR (400 MHz, CDCl3): δ = 8.18 (d, J = 8.0 Hz, 1H), 7.62–7.58 (m, 1H), 7.55–7.51 (m, 1H), 7.40 (d, J = 8.8 Hz, 2H), 7.38 (s, 1H), 7.04 (d, J = 8.8 Hz, 2H), 6.40 (s, 1H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 180.5, 179.8, 161.0, 156.9, 135.2, 135.1, 131.7, 130.8, 130.5, 129.8, 129.7, 128.7, 127.1, 114.3, 55.5; HRMS (ESI): m/z [M + H]+ calcd for C16H10O3: 264.0786; found: 264.0775.
4-(3,4-Dimethoxyphenyl)naphthalene-1,2-dione (5g). The reaction was carried out according to general method A. Eluent, hexane/ethyl acetate = 90
:10 (yellow solid, 96 mg, 50.0% yield, mp 164–165 °C); 1H NMR (400 MHz, CDCl3): δ = 8.20 (d, J = 8.0 Hz, 1H), 7.62–7.58 (m, 1H), 7.55–7.52 (t, 1H), 7.43–7.41 (d, 1H), 7.06–7.04 (q, 1H), 7.01–7.00 (d, 1H), 6.96–6.95 (d, 1H), 6.43 (s, 1H), 3.97 (s, 3H), 3.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 180.5, 179.7, 157.0, 150.5, 149.1, 135.2, 135.1, 131.7, 130.8, 130.6, 129.6, 129.0, 127.1, 121.2, 111.3, 111.2, 56.1, 56.0.
10-Chloro-6-(4-methoxyphenyl)-5H-benzo[a]phenothiazin-5-one (8a). The reaction was carried out according to general method C. Eluent, DCM (red powder, 54.13 mg, 92.0% yield, mp 220–223 °C); 1H NMR (400 MHz, CDCl3): δ = 8.90 (d, J = 7.2 Hz, 1H), 8.35 (d, J = 6.8 Hz, 1H), 7.94 (d, J = 2.0 Hz, 1H), 7.83–7.76 (m, 2H), 7.34–7.24 (m, 4H), 7.08 (d, J = 8.4 Hz, 2H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 178.9, 160.0, 146.2, 139.3, 135.1, 134.3, 132.9, 132.3, 132.1, 131.8, 131.7, 130.6, 129.7, 126.4, 126.2, 125.7, 125.6, 122.3, 114.7, 55.3. HRMS (ESI): m/z [M + H]+ calcd for C23H14ClNO2S: 403.0434; found: 403.0464.
Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
A. K. T. P. and S. P thank the UGC-RFSMS, New Delhi for the award of the fellowship. We thank DST-FIST and DST-PURSE New Delhi, India for NMR and HRMS facilities at the School of Chemistry, Bharathidasan University, Tiruchirappalli, and India.References
- (a) J. L. Bolton and T. Dunlap, Chem. Res. Toxicol., 2017, 30, 13–37 Search PubMed; (b) N. E. Najjar, H. G. Muhtasib, R. A. Ketola, P. Vuorela, A. Urtti and H. Vuorela, Phytochem. Rev., 2011, 10, 353–370 CrossRef.
- (a) M. Sugumaran and V. Semensi, J. Biol. Chem., 1991, 266(10), 6073–6078 CAS; (b) J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS PubMed.
- (a) G. G. Dias, A. King, F. De Moliner, M. Vendrell and E. S. Junior, Chem. Soc. Rev., 2018, 47, 12–27 RSC.
- (a) E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179–11202 RSC and references cited therein.
- (a) M. K. Hadden, S. A. Hill, J. Davenport, R. L. Matts and B. S. Blagg, Bioorg. Med. Chem., 2009, 37, 634 CrossRef PubMed; (b) O. Frutos, C. Atienza and A. M. Echavarren, Eur. J. Org. Chem., 2001, 163–171 CrossRef; (c) C. J. Zheng, M. J. Sohn and W. G. Kim, J. Antibiot., 2006, 59, 808–812 CrossRef CAS PubMed; (d) Y. Zhang, D. Guo, S. Ye, Z. Liu and G. Zhu, Org. Lett., 2017, 19, 1302–1305 CrossRef CAS.
- (a) S. L. Lanny and W. R. Steven, J. Org. Chem., 1993, 58, 408–413 CrossRef; (b) T. Nuria, A. M. Echavarren and M. C. Paredes, J. Org. Chem., 1991, 56, 6490–6494 Search PubMed; (c) X. Gan, W. Jiang, W. Wang and L. Hu, Org. Lett., 2009, 11(3), 589–592 CrossRef CAS PubMed; (d) M. K. Hadden, S. A. Hill, J. Davenport, R. L. Matts and B. S. Blagg, Bioorg. Med. Chem., 2009, 17, 634–640 CrossRef CAS PubMed; (e) Z. Hassan, I. Ullah, I. Ali, R. A. Khera, I. Knepper, A. Ali, T. Patonay, A. Villinger and P. Langer, Tetrahedron, 2013, 69, 460–469 CrossRef CAS; (f) A. R. Louvis, N. A. A. Silva, F. S. Semaan, F. C. Da-Silva, G. Saramago, L. C. de Souza, B. L. A. Ferreira, H. C. Castro, J. P. Salles, A. L. A. Souza, R. X. Faria, V. F. Ferreira and D. L. Martins, New J. Chem., 2016, 40, 7643–7656 RSC.
- (a) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. D. Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS PubMed; (b) D. Wang, B. Ge, L. Du, H. Miao and Y. Ding, Synlett, 2014, 25, 2895–2898 CrossRef CAS; (c) D. Wang, B. Ge, A. Ju, Y. Zhou, C. Xu and Y. Ding, J. Organomet. Chem., 2015, 780, 30–33 CrossRef CAS; (d) S. E. Walker, A. D. Jordan-Hore, J. G. Johnson, S. A. Macgregor and A. L. Lee, Angew. Chem., Int. Ed., 2014, 53, 13876–13879 CrossRef CAS PubMed; (e) A. Ortega, Á. Rincón, K. L. Jiménez-Aliaga, P. Bermejo-Bescós, S. Martín-Aragón, M. T. Molina and A. G. Csákÿ, Bioorg. Med. Chem. Lett., 2011, 21, 2183–2187 CrossRef CAS PubMed; (f) J. Wang, S. Wang, G. Wang, J. Zhang and X. Q. Yu, Chem. Commun., 2012, 48, 11769–11771 RSC; (g) K. Komeyama, T. Kashihara and K. Takaki, Tetrahedron Lett., 2013, 54, 1084–1086 CrossRef CAS; (h) A. Deb, S. Manna, A. Maji, U. Dutta and D. Maiti, Eur. J. Org. Chem., 2013, 5251–5256 CrossRef CAS.
- (a) A. Ilangovan, P. Ashok and G. Satish, Org. Chem. Front., 2015, 2, 1616–1620 RSC; (b) S. Shaaban, A. Jolit, D. Petkova and N. Maulide, Chem. Commun., 2015, 51, 13902–13905 RSC; (c) J. F. Bagli and P. H. Lecuyer, Can. J. Chem., 1961, 39, 1037 CrossRef CAS; (d) A. Honraedt, L. C. François, L. G. Erwan, V. Fernandez and F. F. Xavier, J. Org. Chem., 2013, 78, 4604 CrossRef CAS PubMed; (e) D. Wang, G. Bingyang, L. Liang, J. Shan and Y. Ding, J. Org. Chem., 2014, 79, 8607 CrossRef CAS PubMed; (f) P. Patil, A. Nimonkar and K. G. Akamanchi, J. Org. Chem., 2014, 79, 2331 CrossRef CAS PubMed.
- (a) R. Wang and J. R. Falck, Org. Chem. Front., 2014, 1, 1029–1034 RSC; (b) D. Wang, B. Ge, S. Yang, H. Miao and Y. Ding, Russ. J. Gen. Chem., 2014, 84, 1615–1162 CrossRef CAS; (c) A. Honraedt, F. Le Callonnec, E. Le Grognec, V. Fernandez and F. X. Felpin, J. Org. Chem., 2013, 78(9), 4604–4609 CrossRef CAS PubMed; (d) L. Wang, J. Shen, S. Yang, W. Liu, Q. Chen and M. He, Green Chem., 2018, 20(6), 1290–1296 RSC.
- (a) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117(13), 8787–8863 CrossRef CAS PubMed; (b) T. Itahara, J. Org. Chem., 1985, 50, 5546–5550 CrossRef CAS; (c) S. Zhang, F. Song, D. Zhao and J. You, Chem. Commun., 2013, 49, 4558–4560 RSC; (d) Y. Moon, Y. Jeong, D. Kook and S. Hong, Org. Biomol. Chem., 2015, 13, 3918–3923 RSC; (e) Z. She, Y. Shi, Y. Huang, Y. Cheng, F. Song and J. You, Chem. Commun., 2014, 50(90), 13914–13916 RSC.
- H. X. Dai, G. Li, X. G. Zhang, A. F. Stepan and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed and references cited therein.
- (a) B. Xiao, Y. Fu, J. Xu, T. J. Gong, J. J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468–469 CrossRef CAS PubMed; (b) Z. Huang, L. Jin, Y. Feng, P. Peng, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7151–7155 CrossRef CAS PubMed and references cited therein; (c) Q. Gao, X. Wu, S. Liu and A. Wu, Org. Lett., 2014, 16, 1732–1735 CrossRef CAS PubMed; (d) R. K. Nandi, F. Ratsch, R. Beaud, R. Guillot, C. Kouklovsky and G. Vincent, Chem. Commun., 2016, 52, 5328–5331 RSC; and references cited therein. (e) S. Narute and D. Pappo, Org. Lett., 2017, 19, 2917–2920 CrossRef CAS PubMed and references cited therein.
- (a) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS PubMed; (b) J. C. Xiang, Y. Cheng, M. Wang, Y. D. Wu and A. X. Wu, Org. Lett., 2016, 18, 4360–4363 CrossRef CAS; (c) T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS; (d) A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398–12414 RSC; (e) L. Liu, K. Chen, W. Z. Wu, P. F. Wang, H. Y. Song, H. Sun, X. Wen and Q. L. Xu, Org. Lett., 2017, 19, 3823–3826 CrossRef CAS PubMed.
- Z. Huang and J. P. Lumb, ACS Catal., 2019, 9, 521–555 CrossRef CAS.
- Z. Huang and J. P. Lumb, Angew. Chem., Int. Ed., 2016, 55, 1–6 CrossRef.
- (a) P. Bisel, L. Al-Momani and M. Muller, Org. Biomol. Chem., 2008, 6, 2655–2665 RSC.
- (a) C. H. Sudhakar, R. M. Komal and R. K. Raghava, Chem. Sin., 2017, 8(2), 269–272 CAS; (b) T. Sander, J. Freyss, M. Von-Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed; (c) D. Antoine, M. Olivier and Z. Vincent, Sci. Rep., 2017, 7, 42717 CrossRef PubMed; (d) A. Dahan, J. M. Miller and G. L. Amidon, AAPS J., 2009, 11(4), 740–746 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectral data and copies of spectra. See DOI: 10.1039/c9qo00623k |
| This journal is © the Partner Organisations 2019 |