
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of rigidified shikimic acid derivatives by ring-closing metathesis to imprint inhibitor efficacy against shikimate kinase enzyme†
Marina
Pernas
 a,
Beatriz
Blanco
a,
Emilio
Lence
a,
Paul
Thompson
b,
Alastair R.
Hawkins
b and
Concepción
González-Bello
*a
a,
Beatriz
Blanco
a,
Emilio
Lence
a,
Paul
Thompson
b,
Alastair R.
Hawkins
b and
Concepción
González-Bello
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, Jenaro de la Fuente s/n, 15782 Santiago de Compostela, Spain. E-mail: concepcion.gonzalez.bello@usc.es
bInstitute of Cell and Molecular Biosciences, Medical School, University of Newcastle upon Tyne, Newcastle upon Tyne NE2 4HH, UK
First published on 4th June 2019
Abstract
Diverse rigidified shikimic acids derivatives, which are stable mimetics of the high-energy conformation of shikimic acid, have been synthesized to enhance inhibitor efficacy against shikimate kinase enzyme (SK), an attractive target for antibiotic drug discovery. The synthesis of the reported conformationally restricted shikimic acid derivatives was carried out by ring-closing metathesis of allyloxy vinyl derivatives as the key step. The rigidification of the ligand conformation was used to maximize the effectiveness of the substituents introduced in the ether carbon bridge of the scaffold by pre-orienting their interaction with key residues and enzyme domains that are essential for catalysis and enzyme motion. Molecular Dynamics simulation studies on the enzyme/ligand complexes revealed marked differences in the positioning of the ligand substituent in the active site of the two enzymes studied (SK from Mycobacterium tuberculosis and Helicobacter pylori) and this explains their greater efficacy against one of the enzymes. This enhancement is due to the distinct induced-fit motion of the two homologous enzymes. A 20-fold improvement against the H. pylori enzyme was achieved by the introduction of a CH2OEt group in the rigid ether bridge of the reported shikimic acid analogs.
Introduction
Flexible ligands upon binding to their biological target may suffer an entropic penalty due to the freezing of their rotatable bonds to achieve the active binding conformation.1,2 In some cases these ligands may also adopt high-energy active conformations in order to maximize favorable interactions with the residues involved in the protein binding pocket.3,4 Hence, pre-organization of the ligand conformation or stabilization of the required high-energy active arrangements through the introduction of conformational constraints is a very attractive strategy that is used in drug design.5–9 The rigidification of the ligand conformation can also be considered as an ‘atom-efficient approach’, since it maximizes the efficiency of the functional groups introduced into the initial scaffold during the drug optimization process as the interactions of those groups with the binding pocket are well pre-oriented.10–12We became interested in using this appealing concept in the development of inhibitors of the fifth enzyme of the shikimic acid pathway, namely the shikimate kinase (SK) enzyme. SK is an attractive target for antibiotic drug discovery because (i) it has no counterpart in human cells; and (ii) it is essential in several very relevant pathogenic bacteria that nowadays show high levels of resistance to many antibiotics in clinical use. Specifically, SK is crucial for: (i) Mycobacterium tuberculosis, which is responsible for tuberculosis – a globally established Word Health Organization (WHO) priority; (ii) Helicobacter pylori, which is the causative agent of gastric and duodenal ulcers and has also been classified as a type I carcinogen; and (iii) Pseudomonas aeruginosa, which is one of the most common pathogens in healthcare-associated infections and a WHO critical pathogen for R&D of new antibiotics. SK catalyzes the stereospecific phosphorylation of the C3 hydroxyl group of shikimic acid (1) by transferring the γ-phosphate group of ATP to the hydroxyl group to provide shikimate 3-phosphate and ADP (Fig. 1A). This enzyme is an amazing example of how the specific transformation of only one of three hydroxyl groups of the ligand is achieved by an exquisitely designed stabilization of its high-energy conformation. By forcing the axial disposition of the C4 and C5 hydroxyl groups in 1, the enzyme achieves the equatorial arrangement of the C3 hydroxyl group for selective phosphorylation by ATP.
 | ||
| Fig. 1 A. Schematic representation of shikimic acid (1) recognition by SK. B. Previously reported reversible competitive inhibitors. C. Target compounds. | ||
Based on the aforementioned recognition, we reported previously that the rigidified shikimic acid derivative 2, in which the conformation that the enzyme recognizes for catalysis is fixed by an unsaturated ether bridge between positions C3 and C5 in 1, is a reversible competitive inhibitor of SK from M. tuberculosis (Mt-SK) (Fig. 1B).13 Compound 2 proved to have an inhibition constant (Ki) of 62 μM, which is lower than the enzyme Km (544 μM). The crystal structure of Mt-SK in complex with ADP and 2 (PDB entry 4BQS, 2.15 Å) revealed that the ligand occupies the active site with a similar arrangement and polar interactions (hydrogen bonding and electrostatic interactions) as 1. More importantly, the structure shows that the rigidification of the diaxial conformation of the C4 and C5 hydroxyl groups in 1 by a C3–C5 ether bridge causes a dramatic reduction in the flexibility of the lid and shikimic acid binding (SB) domains, the plasticity of which is essential for catalytic turnover. The SB domain, which involves several highly conserved lipophilic residues, isolates the substrate from the solvent environment to perform the reaction. Molecular Dynamics (MD) simulation studies also revealed that a closed form of the lid and SB domains are required for catalysis.13 Moreover, reduction of the double bond of the C3–C5 ether bridge in 2, to give compound 3, improves the ligand affinity a little more (Ki = 46 μM) by enhancing lipophilic interactions between the ether bridge and residues of the lid domain, thus sealing the active site even more.
Based on these results, we report herein the possible enhancement of the inhibitor efficiency of this scaffold, compound 2, by promoting favorable lipophilic interactions between the ligand and the lid (Fig. 1C). To this end, we carried out the synthesis of rigidified shikimic acid derivatives 4–5 in which the closest sp2 carbon of the unsaturated bridge to the lid was substituted with diverse apolar groups that pre-orient their interaction with this important part of the enzyme. In addition, the relevance of the double bond to ligand affinity was studied with compounds 5. The results of inhibition studies with the SK from M. tuberculosis and from H. pylori, along with MD simulation studies on the enzyme/ligand complexes, allowed us to explain the higher efficacy of the reported compounds observed for the H. pylori enzyme.
Results and discussion
Synthesis of compounds 4–5
The synthesis of conformationally restricted shikimic acid derivatives 4–5 was carried out by ring-closing metathesis of the allyloxy vinyl derivatives 6 as the key step (Fig. 2). | ||
| Fig. 2 Synthetic approach. | ||
In an effort to facilitate the formation of the seven-membered bridge ring in 6, the axial arrangement of the vinyl group in C3 was induced by protecting the hydroxyl groups in the C3 and C4 positions of the shikimic acid derivative as an acetal (Fig. S1†). The key compounds 6 were prepared by Trost allylation of previously reported alcohol 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 using the appropriate allyl methyl carbonates 8.
13 using the appropriate allyl methyl carbonates 8.
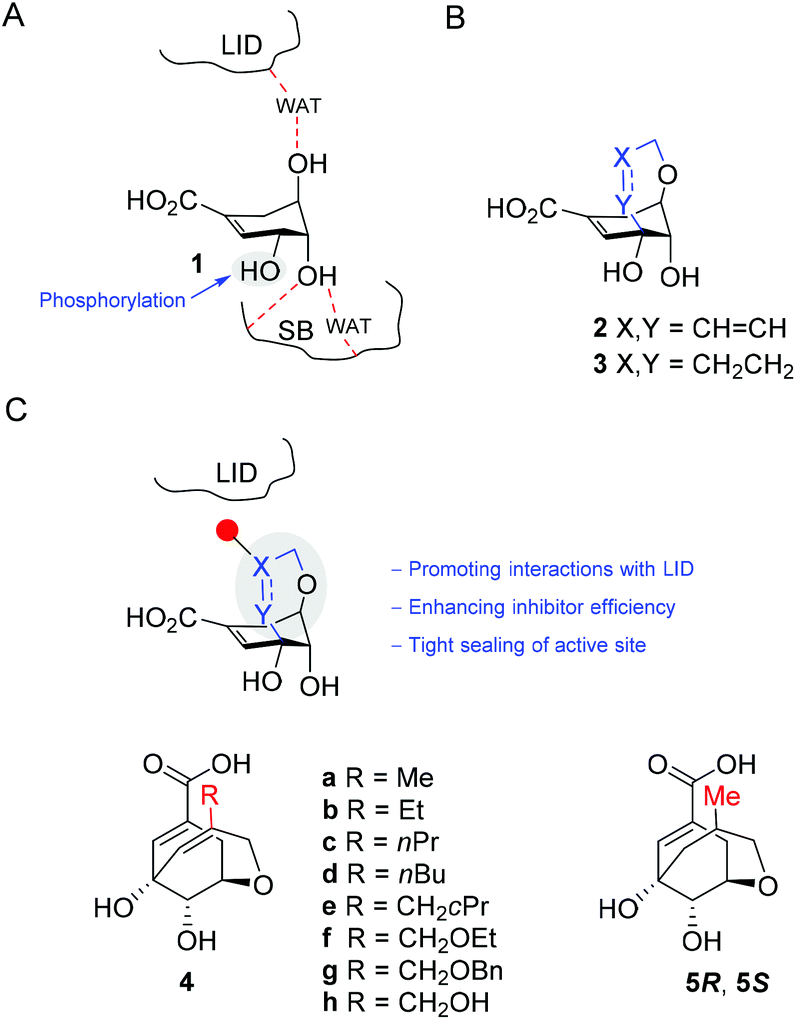
Allyl methyl carbonates 8 were prepared from the corresponding alcohols 14–21 by treatment with methyl chloroformate and pyridine (Scheme 1). Alcohols 15–17 were synthesized by 1,2-reduction of α,β-unsaturated aldehydes 11 (commercially available) and 12–13, with the latter compounds readily prepared from pentanal (9) and hexanal (10), respectively. Alcohol 18 was obtained in four steps from ethyl malonate (22): (i) alkylation of 22 with cyclopropylmethyl bromide; (ii) decarboxylative hydrolysis; (iii) aldol condensation; and (iv) 1,2-reduction of α,β-unsaturated acid 23. Alcohols 20–21 were synthesized by alkylation of commercially available 2-methylene-1,3-propanodiol (19). Finally, carbonate 8i was obtained by TBS-protection of carbonate 8h.
 | ||
| Scheme 1 Synthesis of carbonates 8. Reagents and conditions. (a) HCHO, pyrrolidine, propionic acid, iPrOH, 45 °C. (b) NaBH4, MeOH, Et2O, 0 °C to RT. (c) 1. NaH, DMF, 0 °C; 2. EtBr, RT. (d) 1. NaH, THF, 0 °C; 2. BnBr, RT. (e) MeOCOCl, Py, DCM, 0 °C to RT. (f) 1. NaH, THF, 0 °C; 2. BrCH2cPr, Δ. (g) NaOH (2 M), Δ. (h) piperidine, HCHO, EtOH, 80 °C. (i) BH3·Me2S, THF, 0 °C to RT. (j) TBSCl, DMAP, TBAI, Et3N, DMF, 0 °C to RT. | ||
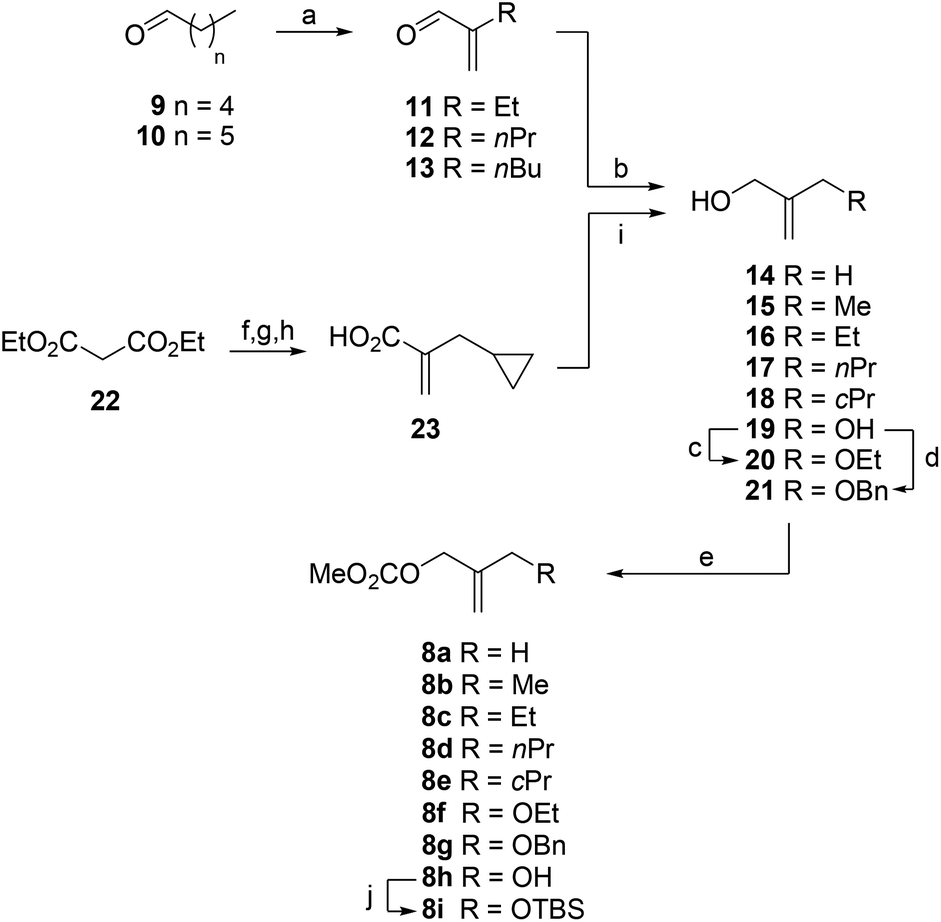
Palladium-catalyzed Trost allylation of 7 with allyl methyl carbonates 8a–g and 8i gave the key intermediates 6a–g and 6i in yields ranging from 32–88% (Scheme 2 and Table 1). Ring-closing metathesis of 6a–g and 6i was achieved by using second-generation Grubbs’ catalyst in toluene at 90 °C to afford bicyclic derivatives 24a–g and 24i in yields ranging from 16–63%, and from 42–99% considering the recovered starting material. Bicyclic derivative 24h was efficiently prepared from 24i by TBS-deprotection with TBAF. As expected, the metathesis reaction proved to be quite sensitive to the presence of substitution in the allyl moiety, since: (i) when R = H the transformation took place at room temperature and in a higher yield (88%);13 and (ii) an increase in the steric hindrance of the substituent led to lower reaction yields and required higher reaction temperatures (Table 1). Derivatives 24g and 24f, which contained a CH2OBn and a CH2OEt group, respectively, gave the lowest yields. These compounds were alternatively prepared by alkylation of alcohol 24h in 64% and 31% (57%) yield, respectively. Deprotection of the acetal group in 24, followed by basic hydrolysis of the resulting esters 25a–h and subsequent protonation with Amberlite IR-120 (H+) ion-exchange resin efficiently afforded the target compounds 4. Finally, compounds 5, which have a flexible substituted ether bridge, were synthesized from methyl derivative 25a by catalytic hydrogenation using Rosenmund catalyst in the presence of pyridine, followed by hydrolysis of the methyl ester to give a 1:1 mixture of epimers in the C4 position, i.e., compounds 5S and 5R, which were separated by HPLC. The configuration of the new chiral center was determined by NOE experiments. Inversion of H10 in bicycles 5S and 5R led to enhancement of the signals for H4 (3.6%) and the methyl group (5.2%), respectively.
 | ||
| Scheme 2 Synthesis of compounds 4–5. Reagents and conditions. (a) 8a–g and 8i, Pd2(dba)3 (cat), dppb, THF, Δ. (b) 2nd generation Grubbs’ catalyst, PhMe, 90 °C. (c) MeOH, HCl (6 M), 60 °C. (d) TBAF, THF, 0 °C. (e) 1. NaH, THF, 0 °C; 2. BnBr, RT. (f) 1. NaH, DMF, 0 °C; 2. EtBr, RT. (g) 1. LiOH (aq.), THF, RT. 2. Amberlite IR-120 (H+), RT. (h) H2, Rosenmund catalyst, MeOH, Py. RT. | ||
Inhibition studies
The inhibitory activity of the reported conformationally restricted shikimic acid derivatives 2–5 was assayed against SK from Helicobacter pylori (Hp-SK) and from M. tuberculosis (Mt-SK). All of the compounds proved to be competitive reversible inhibitors of shikimic acid for both enzymes. The inhibition data (Ki), which were obtained from Dixon plots (1/v vs. [I]), are summarized in Table 2.| Entry | Comp | R | H. pylori | M. tuberculosis |
|---|---|---|---|---|
| a Assay conditions: Tris·HCl (100 mM, pH 7.7), ATP (2.5 mM), NADH (0.2 mM), PEP (1 mM), MgCl2 (5 mM), KCl (100 mM), lactate dehydrogenase/pyruvate kinase (∼2.8 units), 25 °C. For Mt-SK: Km (1) = 544 ± 14 μM; kcat = 295 ± 8 s−1. For Hp-SK: Km (1) = 39 ± 8 μM; kcat = 116 ± 4 ms−1. ND = not determined. | ||||
| 1 | 2 | H | 104 ± 4 | 62 ± 1 (ref. 13) |
| 2 | 3 | — | 47 ± 6 | 46 ± 2 (ref. 13) |
| 3 | 4a | Me | 54.5 ± 5.7 | 28 ± 1 |
| 4 | 4b | Et | 15.5 ± 1.1 | 41 ± 2 |
| 5 | 4c | nPr | 9.2 ± 1.0 | 72 ± 4 |
| 6 | 4d | nBu | 12 ± 2 | 177 ± 3 |
| 7 | 4e | cPr | 10.0 ± 0.6 | 101 ± 2 |
| 8 | 4f | CH2OEt | 5.0 ± 0.3 | 170 ± 3 |
| 9 | 4g | CH2OBn | 68 ± 3 | 121 ± 5 |
| 10 | 4h | CH2OH | 38 ± 3 | 333 ± 10 |
| 11 | 5S | Me | 465 ± 41 | 360 ± 7 |
| 12 | 5R | Me | ND | 645 ± 16 |
In general: (i) the ligands proved to be more potent against the H. pylori enzyme than the M. tuberculosis enzyme; (ii) a rigid ether bridge between the C3 and C5 positions of shikimic acid provided more potent inhibitors (Table 2, entries 3 vs. 11); (iii) for Hp-SK, the inhibition potency of the ligands increased with the length of the substituent chain (Table 2, entries 5 and 8 vs. 1 and 4), while for Mt-SK only the introduction of a methyl group in the rigid ether bridge improved the inhibitory activity (Table 2, entries 3 vs. 5); (iv) the presence of a hydroxyl or an ether group in the substituent only enhanced the inhibitory activity for Hp-SK (Table 2, entries 8 vs. 5).
For the H. pylori enzyme, the best inhibitor in the series was compound 4f, which has a CH2OEt substituent in the ether bridge. This enhanced the inhibitory potency by up to 20-fold. For the M. tuberculosis enzyme, a 2-fold improvement in activity was achieved with compound 4a, which has a methyl group. Computational studies were performed in an effort to gain a better understanding at the atomic level of the differences observed experimentally in the inhibitory potency of the reported conformationally rigid shikimic acid analogs 4–5. The results of these studies are discussed below.
Computational studies
Molecular docking using the GOLD 5.2.214 program and the protein coordinates found in the crystal structures of Hp-SK in complex with shikimate-3-phosphate and ADP (PDB entry 3MUF,15 2.3 Å) and of Mt-SK in complex with 2 and ADP (PDB entry 4BQS,13 2.15 Å) were carried out first. The highest score solutions obtained by docking were further analyzed by Molecular Dynamics (MD) simulation studies in order to assess the stability and therefore the reliability of the postulated binding. The monomer of the Hp-SK/ATP/Mg2+/ligand and Mt-SK/ATP/Mg2+/ligand complexes in a truncated octahedron of water molecules obtained with the molecular mechanics force field AMBER16 was employed and the system was then subjected to 100 ns of dynamic simulation. The latter was carried out with the most active ligands, compounds 4a–d and 4f, as well as the analogs with a flexible ether bridge, i.e., 5S and 5R (Fig. 3).
 | ||
| Fig. 3 Binding mode of compounds 4a (green), 4c (cyan) and 4f (yellow) obtained by docking and MD simulation studies in the active site of the Hp-SK (gray) and Mt-SK (dark blue) enzymes. A–C. Overall view of the Hp-SK/ATP/Mg2+/ligand complexes. Snapshots after 80 ns of simulation are shown. Mg2+ (sphere) and ligands (sticks) and ATP (sticks) are displayed. D–F. Close view of ligands 4a, 4c and 4f in the SB binding pocket of Hp-SK. Note how these ligands and the substituents of the ether bridge are perfectly surrounded by the enzyme, in particular for compounds 4c and 4f. G–H. Close view of ligands 4a and 4f in the Mt-SK/ATP/Mg2+/ligand complexes. Note how for ligand 4f the substituent of the ether bridge would not be embedded in the active site as for the H. pylori enzyme. I. Percentage of conformations of the ligands 4b–d and 4f during the 100 ns of simulation in which the substituent of the ether bridge (Et, nPr, nBu, CH2OEt) would be located pointing towards the active site (inside). | ||
The results of the computational studies revealed that, in all cases, the ligands would be stable in the shikimic acid active site, since significant variations were not observed during the whole simulation, both in the position of the ligand and in the protein backbone (Fig. S2 and S3†). As one would expect, the ligands would be anchored to the active site by the same electrostatic and polar interactions as the original compound 2 (Fig. S4†). More importantly, relevant differences were identified in the arrangement of the substituent of the ether bridge of the ligands for both enzymes and this would explain the experimentally obtained activity. Thus, for Hp-SK and during most of the simulation, these substituents were mainly embedded in the active site, with both the lid and the SB domain completely surrounding the entire ligand (Fig. 3A–F). For compounds 4b–d and 4f (R ≠ H, Me), the percentage of conformations with the substituent ‘inside’ the active site increased as the chain length increased, which is in good agreement with the observed improvement in the inhibitory potency (CH2OEt > nBu > nPr > Et) (Fig. 3I). These values were calculated by analyzing the variation of the dihedral angle between the atoms C5 (CAF), C4 (CAE) and the first two atoms of the substituent, C (CAP) and C (CAR)/O (OAR), in 4b–d and 4f during the whole simulation (Fig. S5 and S6†). ‘Substituent inside conformations’ were considered for values of the dihedral angle between −50° and −150°. As a result of this arrangement, the shikimic acid active site remained neatly closed, thus avoiding the entrance of the natural substrate, because the ligands caused a dramatic reduction in the flexibility of the lid and SB domain by a series of favorable apolar interactions between the substituent and the residues in this pocket (Fig. 4A–D). It is worth highlighting that MD studies in the enzyme product complex, i.e., in the presence of ADP and shikimate-3-phosphate, revealed that the flexibility of the lid and the SB domain are key for the catalytic turnover.13 The lid is the substrate-covering loop that closes over the shikimic acid binding site for catalysis and it contains the essential residue Arg116/Arg117 (H. pylori and M. tuberculosis, respectively). NMR studies revealed that this residue might also be involved in the phosphoryl-transfer mechanism catalyzed by SK by activating and positioning the reaction intermediate for subsequent nucleophilic attack by the C3 hydroxyl group in 1.18 The aforementioned apolar interactions would be more numerous as the length of the chain increases, which would explain the enhancement in ligand affinity. In general, these interactions would involve the residues of the: (i) lid: Arg116 (essential), Pro117 (conserved) and Leu118; (ii) the SB domain: Val44 and Arg45; and (iii) the P-loop: Met10 (Fig. S7†). For the most potent inhibitor, compound 4f, an additional interaction was identified between the oxygen atom of the substituent and the amide main chain (carbonyl) of Val44 through a water network, and this could explain the higher affinity of 4f for Hp-SK than 4d, which has a CH2 group in the same position (Fig. 4D and C, respectively).
 | ||
| Fig. 4 Detailed view of the interactions of the ether bridge substituent in 4a, 4c–d and 4f with Hp-SK (A–D) and in 4a and 4f with Mt-SK (E–F) in their respective enzyme complexes. Relevant side chain residues are shown and labeled. Apolar (magenta) and polar (blue) contacts are shown as dashed lines. The interaction of the oxygen atom of the substituent in 4f with the amide main chain (carbonyl) of Val44 through a water network is highlighted with blue shading. | ||
Moreover, for ligands 5, the simulation studies revealed a different behavior of both compounds, mainly relative to the ether bridge. Thus, while for ligand 5R no significant conformational changes were observed during the dynamic simulation, this was not the case for ligand 5S. The ether bridge moiety in 5S underwent a conformational change to locate the methyl group in parallel to the cyclohexene ring. This conformation remained stable after ∼40 ns of simulation (Fig. S8†). As a consequence, for both ligands 5, an interaction of the methyl group with the carbon chain of the essential arginine was not identified, as observed with compound 4f and previously reported saturated derivative 3. This fact revealed how the rigidity of the ether bridge in the ligands would be crucial to fix the position and direction of the substituent towards the key residues of the lid.
In contrast to the above, for Mt-SK, as the length of the substituent increases (R = Et, nPr, nBu, CH2OEt) the ligands would be located preferentially with the substituent pointing outside the active site (Fig. 3G–H). In this arrangement, the introduction of this type of substituent in 2 would not contribute to an improvement in ligand affinity since additional interactions with the residues of the active site could not be established (Fig. 4E–F).
For both enzymes, the substituent of the ligand interacts with a similar region of the lid and this is quite similar in terms of amino acid sequences. However, the results of the computational studies revealed a clear and markedly distinct induced fitting of the ligands by the two enzymes, which would explain the differences found. These are due to key differences in the folding of the lid over the active site – a situation that can be easily visualized by analysis of the vibrational modes of the two enzymes (Fig. 5).19
 | ||
| Fig. 5 Overall view of the Hp-SK and Mt-SK motion obtained by examination of the vibrational modes. The motion of the lid is highlighted with blue and yellow shading, respectively. Note how the folding of the lid and as a consequence the essential arginine that it contains is quite distinct in the two enzymes. | ||
Conclusions
The functionalization of the double bond of previously identified scaffold 2, a stable mimetic of the high-energy conformation of shikimic acid and a competitive reversible inhibitor of the shikimate kinase enzyme, was carried out by ring-closing metathesis of allyloxy vinyl derivatives 6 as the key step. The latter compounds were prepared by Trost allylation of previously reported alcohol 7 with allyl methyl carbonates 8. The RCM approach required reaction temperatures of 90 °C and the use of second-generation Grubbs’ catalyst.The results obtained for the rigidified shikimic acid derivatives reported, namely compounds 4–5, with the two enzymes studied, SK from M. tuberculosis and from H. pylori, revealed that: (i) the rigidification of the functionalized ether bridge between C3 and C5 of the shikimic acid is crucial for improving ligand affinity; (ii) this functionalization generally provides more potent analogs against the H. pylori enzyme than the M. tuberculosis enzyme. A 20-fold improvement against the H. pylori enzyme was achieved by the introduction of a CH2OEt group in the rigid ether bridge of the reported shikimic acid analogs. For the M. tuberculosis enzyme, the introduction of a methyl group in 2 enhanced the ligand potency by a factor of two.
Computational studies revealed that the differences in affinity found with the two homologous enzymes are due to the distinct induced-fit conformation adopted by the two enzymes upon ligand binding, which mainly involves the substrate-covering loop (lid). For Hp-SK, the substituents (R = Et, nPr, nBu, CH2OEt) would be embedded in the active site, with both the lid and the SB domain completely surrounding the entire ligand. As a result, the active site would be neatly closed because the ligands cause a dramatic reduction in the flexibility of the lid and SB domain through a series of favorable apolar interactions between the substituent and the residues in this pocket. In contrast, for Mt-SK, as the length of the substituent increases (R = Et, nPr, nBu, CH2OEt) this moiety of the ligand would be pointing away from the active site and therefore they would not contribute to an improvement in ligand affinity. The results reported here can be considered as a good example of how the rigidification of a ligand is a useful strategy to enhance ligand affinity for a target due to the pre-orientation and maximization of the interactions of its substituents.
Experimental
General
All starting materials and reagents were commercially available and were used without further purification. 1H NMR spectra (250, 300 and 500 MHz) and 13C NMR spectra (63, 75 and 125 MHz) were measured in deuterated solvents. J values are given in hertz. NMR assignments were carried out by a combination of 1 D, COSY, and DEPT-135 experiments. FT-IR spectra were recorded in a PerkinElmer two FTIR spectrometer with attenuated total reference. [α]20D = values are given in 10−1 deg cm2 g−1. MilliQ deionized water was used in all the buffers. Melting points were measured in a Büchi M-560 apparatus. The experimental procedures for the synthesis of carbonates 8 are described in the ESI.†General procedure for synthesis of compounds 6
To a stirred solution of Pd2(dba)3 (0.025 mmol) and dppb (0.1 mmol) in dry THF (0.8 mL), under argon and at room temperature, were added the alcohol 713 (1 mmol), followed by a solution of the carbonates 8a–g and 8i (1.5 mmol) in dry THF (3 mL, 0.5 M). The resulting suspension was heated at 60 °C for 24 h and then cooled to room temperature. The mixture was filtered over Celite® and the residue was washed with diethyl ether. The filtrate and washings were evaporated under reduced pressure to yield an oil which was purified by flash chromatography to afford the allyl ethers 6.
:70) diethyl ether/hexane. Yield = 88% (52 mg). Colorless oil. [α]20D = +107.6° (c 3.5, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.64 (m, 1H, H2), 5.92 (dd, J = 17.3 and 10.7 Hz, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.44 (dd, J = 17.3 and 1.5 Hz, 1H, CHCH2), 5.17 (dd, J = 10.7 and 1.5 Hz, 1H, CHCH2), 4.89 (d, J = 18.6 Hz, 2H, H3CCHCH2), 4.09 (d, J = 4.0 Hz, 1H, H4), 4.02 (dd, J = 6.6 and 3.9 Hz, 1H, H5), 3.91 (d, J = 5.3 Hz, 2H, CH2), 3.76 (s, 3H, OCH3), 2.64 (dd, J = 18.0 and 2.6 Hz, 1H, CHH-6), 2.44 (ddd, J = 18.0, 3.9 and 2.7 Hz, 1H, CHH-6), 1.69 (s, 3H, CH3), 1.41 (s, 3H, CH3) and 1.34 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.4 (C), 141.9 (C), 137.8 (C), 137.6 (C), 126.2 (C), 115.0 (CH2), 112.1 (CH2), 109.1 (C), 80.1 (C), 77.3 (CH), 73.3 (CH2), 73.2 (CH), 51.9 (CH), 27.9 (CH3), 26.9 (CH3), 24.3 (CH2) and 19.4 (CH3) ppm. FTIR (film) ν: 1719 (CO) cm−1. MS (ESI) m/z = 331 (MNa+). HRMS calcd for C17H24O5Na (MNa+): 331.1516; found, 331.1511.
:50) diethyl ether/hexane. Yield = 32% (213 mg). It was recovered 221 mg of starting material. Corrected yield = 74%. Colorless oil. [α]20D = +99.1° (c 1.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.64 (t, J = 1.3 Hz, 1H, H2), 5.92 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.45 (dd, J = 1.5 and 17.3 Hz, 1H, CHCHH), 5.18 (dd, J = 1.4 and 10.7 Hz, 1H, CHCHH), 4.95 (m, 1H, CCHH), 4.87 (m, 1H, CCHH), 4.09 (m, 1H, H5), 4.02 (m, 1H, H4), 3.99 (d, J = 12.5 Hz, 1H, OCHH), 3.92 (d, J = 12.5 Hz, 1H, OCHH), 3.76 (s, 3H, OCH3), 2.65 (dd, J = 2.5 and 17.9 Hz, 1H, CHH-6), 2.44 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 2.02 (q, J = 7.4 Hz, 2H, CH2CH3), 1.42 (s, 3H, CH3), 1.34 (s, 3H, CH3) and 1.02 (t, J = 7.4 Hz, 3H, CH2CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 147.5 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.1 (CH2), 110.3 (CH2), 109.1 (C), 80.2 (C), 77.3 (CH), 73.2 (CH), 72.5 (CH2), 52.0 (OCH3), 28.0 (CH3), 27.0 (CH3), 25.9 (CH2), 24.4 (CH2) and 12.0 (CH3) ppm. FTIR (film): 1710 (CO) cm−1. MS (ESI) m/z = 345 (MNa+). HRMS calcd for C18H26O5Na (MNa+): 345.1672; found, 345.1681.
:80) diethyl ether/hexane. Yield = 47% (122 mg). Colorless oil. [α]20D = +92.8° (c 1.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.62 (t, J = 1.4 Hz, 1H, H2), 5.89 (dd, J = 10.7 and 17.4 Hz, 1H, CHCH2), 5.42 (dd, J = 1.5 and 17.3 Hz, 1H, CHCHH), 5.14 (dd, J = 1.5 and 10.7 Hz, 1H, CHCHH), 4.93 (br s, 1H, CCHH), 4.83 (br s, 1H, CCHH), 4.06 (br d, J = 4.0 Hz, 1H, H4), 4.00 (td, J = 2.6 and 3.9 Hz, 1H, H5), 3.94 (d, J = 12.6 Hz, 1H, OCHH), 3.87 (d, J = 12.6 Hz, 1H, OCHH), 3.73 (s, 3H, OCH3), 2.63 (dd, J = 2.6 and 18.0 Hz, 1H, CHH-6), 2.41 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 1.95 (t, J = 7.5 Hz, 2H, CH2CH3), 1.47–1.35 (m, 5H, CH2 + CH3), 1.31 (s, 3H, CH3) and 0.86 (t, J = 7.3 Hz, 3H, CH2CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 145.8 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.0 (CH2), 111.4 (CH2), 109.1 (C), 80.2 (C), 77.4 (CH), 73.3 (CH), 72.3 (CH2), 52.0 (OCH3), 35.2 (CH2), 28.0 (CH3), 27.0 (CH3), 24.4 (CH2), 20.8 (CH2) and 13.9 (CH3) ppm. FTIR (film): 1711 (CO) cm−1. MS (ESI) m/z = 359 (MNa+). HRMS calcd for C19H28O5Na (MNa+): 359.1829; found, 359.1826.
:90) diethyl ether/hexane. Yield = 49% (250 mg). Colorless oil. [α]20D = 105.9° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.64 (m, 1H, H2), 5.91 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.44 (dd, J = 1.5 and 17.3 Hz, 1H, CHCHH), 5.17 (dd, J = 1.5 and 10.7 Hz, 1H, CHCHH), 4.94 (m, 1H, CCHH), 4.85 (m, 1H, CCHH), 4.08 (td, J = 1.1 and 4.0 Hz, 1H, H4), 4.01 (dt, J = 2.5 and 3.8 Hz, 1H, H5), 3.96 (d, J = 12.8 Hz, 1H, OCHH), 3.89 (d, J = 12.8 Hz, 1H, OCHH), 3.75 (s, 3H, OCH3), 2.64 (m, 1H, CHH-6), 2.43 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 1.99 (t, J = 6.9 Hz, 2H, CH2(CH2)2CH3), 1.43–1.23 (m, 4H, CH2(CH2)2CH3), 1.41 (s, 3H, CH3), 1.33 (s, 3H, CH3) and 0.88 (t, J = 7.1 Hz, 3H, CH2(CH2)2CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 146.1 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.0 (CH2), 111.2 (CH2), 109.1 (C), 80.2 (C), 77.3 (CH), 73.2 (CH), 72.4 (CH2), 52.0 (OCH3), 32.8 (CH2), 29.8 (CH2), 28.0 (CH3), 27.0 (CH3), 24.4 (CH2), 22.5 (CH2) and 14.0 (CH3) ppm. FTIR (film): 1717 (CO) cm−1. MS (ESI) m/z = 373 (MNa+). HRMS calcd for C20H30O5Na (MNa+): 373.1985; found, 373.1982.
:80) diethyl ether/hexane. Yield = 34% (89 mg). Colorless oil. [α]20D = +93.8° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.64 (m, 1H, H2), 5.91 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.44 (dd, J = 1.4 and 17.3 Hz, 1H, CHCHH), 5.18 (dd, J = 1.4 and 10.7 Hz, 1H, CHCHH), 5.01 (br s, 1H, CCHH), 4.97 (br s, 1H, CCHH), 4.07 (m, 1H, H5), 4.04–4.02 (m, 2H, H4 + OCHH), 3.95 (br d, J = 12.5 Hz, 1H, OCHH), 3.76 (s, 3H, OCH3), 2.65 (dd, J = 2.5 and 18.0 Hz, 1H, CHH-6), 2.44 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 1.90 (d, J = 6.8 Hz, 2H, CH2CH(CH2)2), 1.42 (s, 3H, CH3), 1.34 (s, 3H, CH3), 0.84–0.70 (m, 1H, CH2CH(CH2)2), 0.50–0.44 (m, 2H, CH2CHCH2CH2) and 0.08–0.03 (m, 2H, CH2CHCH2CH2) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.6 (C), 146.1 (C), 138.0 (CH), 137.8 (CH), 126.4 (C), 115.2 (CH2), 111.6 (CH2), 109.2 (C), 80.3 (C), 77.5 (CH), 73.3 (CH), 72.6 (CH2), 52.1 (OCH3), 38.2 (CH2), 28.1 (CH3), 27.1 (CH3), 24.4 (CH2), 9.3 (CH), 4.8 (CH2) and 4.7 (CH2) ppm. FTIR (film): 1717 (CO) cm−1. MS (ESI) m/z = 371 (MNa+). HRMS calcd for C20H28O5Na (MNa+): 371.1829; found, 371.1825.
:80) diethyl ether/hexane. Yield = 54% (190 mg). Colorless oil. [α]20D = +85.2° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.63 (t, J = 1.2 Hz, 1H, H2), 5.89 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.43 (dd, J = 1.4 and 17.3 Hz, 1H, CHCHH), 5.16 (dd, J = 1.4 and 10.7 Hz, 1H, CHCHH), 5.12 (m, 2H, CCH2), 4.08–3.96 (m, 4H, H4 + H5 + OCH2), 3.91 (br s, 2H, CH2OEt), 3.74 (s, 3H, OCH3), 3.44 (q, J = 7.0 Hz, 2H, OCH2CH3), 2.64 (dd, J = 2.5 and 18.0 Hz, 1H, CHH-6), 2.43 (ddd, J = 2.8, 3.7 and 18.0 Hz, 1H, CHH-6), 1.40 (s, 3H, CH3), 1.32 (s, 3H, CH3) and 1.18 (t, J = 7.0 Hz, 3H, OCH2CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 142.9 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.1 (CH2), 113.9 (CH2), 109.2 (C), 80.2 (C), 77.4 (CH), 73.7 (CH), 71.4 (CH2), 70.3 (CH2), 65.9 (CH2), 52.0 (OCH3), 28.0 (CH3), 27.0 (CH3), 24.5 (CH2) and 15.3 (CH3) ppm. FTIR (film): 1716 (CO) cm−1. MS (ESI) m/z = 375 (MNa+). HRMS calcd for C19H28O6Na (MNa+): 375.1778; found, 375.1777.
:50) diethyl ether/hexane. Yield = 63% (207 mg). Yellow oil. [α]20D = +85.9° (c 0.8, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 7.38–7.28 (m, 5H, 5 × ArH), 6.65 (t, J = 1.3 Hz, 1H, H2), 5.89 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.43 (dd, J = 1.4 and 17.3 Hz, 1H, CHCHH), 5.18–5.14 (m, 3H, CHCHH + CCH2), 4.49 (s, 2H, OCH2), 4.12–4.02 (m, 4H, H4 + H5 + CH2OBn), 3.99 (br s, 2H, OCH2Ph), 3.76 (s, 3H, OCH3), 2.65 (dd, J = 2.5 and 18.0 Hz, 1H, CHH-6), 2.45 (ddd, J = 2.8, 3.7 and 18.0 Hz, 1H, CHH-6), 1.42 (s, 3H, CH3) and 1.34 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 142.6 (C), 138.3 (C), 137.9 (CH), 137.7 (CH), 128.5 (2 × CH), 127.8 (2 × CH), 127.8 (CH), 126.3 (C), 115.2 (CH2), 114.3 (CH2), 109.2 (C), 80.2 (C), 77.4 (CH), 73.7 (CH), 72.3 (CH2), 70.9 (CH2), 70.3 (CH2), 52.0 (OCH3), 28.0 (CH3), 27.0 (CH3) and 24.5 (CH2) ppm. FTIR (film): 1709 (CO) cm−1. MS (ESI) m/z = 437 (MNa+). HRMS calcd for C24H30O6Na (MNa+): 437.1935; found, 437.1936.
:75) diethyl ether/hexane. Yield = 43% (72 mg). Colorless oil. [α]20D = +63.2° (c 1.0, CHCl3). 1H NMR (250 MHz, CDCl3) δ: 6.63 (br s, 1H, H2), 5.89 (dd, J = 10.7 and 17.3 Hz, 1H, HCCH2), 5.43 (dd, J = 1.4 and 17.3 Hz, 1H, HCCHH), 5.18–5.14 (m, 2H, HCCHH + CCHH), 5.04 (br s, 1H, CCHH), 4.10 (br s, 2H, OCH2), 4.07–3.94 (m, 4H, H4 + H5 + CH2OTBS), 3.75 (s, 3H, OCH3), 2.63 (dd, J = 1.9 and 18.0 Hz, 1H, CHH-6), 2.42 (dt, J = 3.3 and 18.0 Hz, 1H, CHH-6), 1.40 (s, 3H, CH3), 1.33 (s, 3H, CH3), 0.89 (s, 9H, C(CH3)3) and 0.04 (s, 6H, 2 × CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 145.0 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.2 (CH2), 112.0 (CH2), 109.2 (C), 80.2 (C), 77.3 (CH), 73.3 (CH), 70.2 (CH2), 63.9 (CH2), 52.1 (OCH3), 28.0 (CH3), 27.0 (CH3), 26.0 (C(CH3)3), 24.4 (CH2), 18.5 (C(CH3)3) and −5.3 (2 × CH3) ppm. FTIR (film): 1716 (CO) cm−1. MS (ESI) m/z = 461 (MNa+). HRMS calcd for C23H38O6SiNa (MNa+): 461.2330; found, 461.2330.
CH2), 5.44 (dd, J = 17.3 and 1.5 Hz, 1H, CHCH2), 5.17 (dd, J = 10.7 and 1.5 Hz, 1H, CHCH2), 4.89 (d, J = 18.6 Hz, 2H, H3CCHCH2), 4.09 (d, J = 4.0 Hz, 1H, H4), 4.02 (dd, J = 6.6 and 3.9 Hz, 1H, H5), 3.91 (d, J = 5.3 Hz, 2H, CH2), 3.76 (s, 3H, OCH3), 2.64 (dd, J = 18.0 and 2.6 Hz, 1H, CHH-6), 2.44 (ddd, J = 18.0, 3.9 and 2.7 Hz, 1H, CHH-6), 1.69 (s, 3H, CH3), 1.41 (s, 3H, CH3) and 1.34 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.4 (C), 141.9 (C), 137.8 (C), 137.6 (C), 126.2 (C), 115.0 (CH2), 112.1 (CH2), 109.1 (C), 80.1 (C), 77.3 (CH), 73.3 (CH2), 73.2 (CH), 51.9 (CH), 27.9 (CH3), 26.9 (CH3), 24.3 (CH2) and 19.4 (CH3) ppm. FTIR (film) ν: 1719 (CO) cm−1. MS (ESI) m/z = 331 (MNa+). HRMS calcd for C17H24O5Na (MNa+): 331.1516; found, 331.1511.
:50) diethyl ether/hexane. Yield = 32% (213 mg). It was recovered 221 mg of starting material. Corrected yield = 74%. Colorless oil. [α]20D = +99.1° (c 1.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.64 (t, J = 1.3 Hz, 1H, H2), 5.92 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.45 (dd, J = 1.5 and 17.3 Hz, 1H, CHCHH), 5.18 (dd, J = 1.4 and 10.7 Hz, 1H, CHCHH), 4.95 (m, 1H, CCHH), 4.87 (m, 1H, CCHH), 4.09 (m, 1H, H5), 4.02 (m, 1H, H4), 3.99 (d, J = 12.5 Hz, 1H, OCHH), 3.92 (d, J = 12.5 Hz, 1H, OCHH), 3.76 (s, 3H, OCH3), 2.65 (dd, J = 2.5 and 17.9 Hz, 1H, CHH-6), 2.44 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 2.02 (q, J = 7.4 Hz, 2H, CH2CH3), 1.42 (s, 3H, CH3), 1.34 (s, 3H, CH3) and 1.02 (t, J = 7.4 Hz, 3H, CH2CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 147.5 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.1 (CH2), 110.3 (CH2), 109.1 (C), 80.2 (C), 77.3 (CH), 73.2 (CH), 72.5 (CH2), 52.0 (OCH3), 28.0 (CH3), 27.0 (CH3), 25.9 (CH2), 24.4 (CH2) and 12.0 (CH3) ppm. FTIR (film): 1710 (CO) cm−1. MS (ESI) m/z = 345 (MNa+). HRMS calcd for C18H26O5Na (MNa+): 345.1672; found, 345.1681.
:80) diethyl ether/hexane. Yield = 47% (122 mg). Colorless oil. [α]20D = +92.8° (c 1.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.62 (t, J = 1.4 Hz, 1H, H2), 5.89 (dd, J = 10.7 and 17.4 Hz, 1H, CHCH2), 5.42 (dd, J = 1.5 and 17.3 Hz, 1H, CHCHH), 5.14 (dd, J = 1.5 and 10.7 Hz, 1H, CHCHH), 4.93 (br s, 1H, CCHH), 4.83 (br s, 1H, CCHH), 4.06 (br d, J = 4.0 Hz, 1H, H4), 4.00 (td, J = 2.6 and 3.9 Hz, 1H, H5), 3.94 (d, J = 12.6 Hz, 1H, OCHH), 3.87 (d, J = 12.6 Hz, 1H, OCHH), 3.73 (s, 3H, OCH3), 2.63 (dd, J = 2.6 and 18.0 Hz, 1H, CHH-6), 2.41 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 1.95 (t, J = 7.5 Hz, 2H, CH2CH3), 1.47–1.35 (m, 5H, CH2 + CH3), 1.31 (s, 3H, CH3) and 0.86 (t, J = 7.3 Hz, 3H, CH2CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 145.8 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.0 (CH2), 111.4 (CH2), 109.1 (C), 80.2 (C), 77.4 (CH), 73.3 (CH), 72.3 (CH2), 52.0 (OCH3), 35.2 (CH2), 28.0 (CH3), 27.0 (CH3), 24.4 (CH2), 20.8 (CH2) and 13.9 (CH3) ppm. FTIR (film): 1711 (CO) cm−1. MS (ESI) m/z = 359 (MNa+). HRMS calcd for C19H28O5Na (MNa+): 359.1829; found, 359.1826.
:90) diethyl ether/hexane. Yield = 49% (250 mg). Colorless oil. [α]20D = 105.9° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.64 (m, 1H, H2), 5.91 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.44 (dd, J = 1.5 and 17.3 Hz, 1H, CHCHH), 5.17 (dd, J = 1.5 and 10.7 Hz, 1H, CHCHH), 4.94 (m, 1H, CCHH), 4.85 (m, 1H, CCHH), 4.08 (td, J = 1.1 and 4.0 Hz, 1H, H4), 4.01 (dt, J = 2.5 and 3.8 Hz, 1H, H5), 3.96 (d, J = 12.8 Hz, 1H, OCHH), 3.89 (d, J = 12.8 Hz, 1H, OCHH), 3.75 (s, 3H, OCH3), 2.64 (m, 1H, CHH-6), 2.43 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 1.99 (t, J = 6.9 Hz, 2H, CH2(CH2)2CH3), 1.43–1.23 (m, 4H, CH2(CH2)2CH3), 1.41 (s, 3H, CH3), 1.33 (s, 3H, CH3) and 0.88 (t, J = 7.1 Hz, 3H, CH2(CH2)2CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 146.1 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.0 (CH2), 111.2 (CH2), 109.1 (C), 80.2 (C), 77.3 (CH), 73.2 (CH), 72.4 (CH2), 52.0 (OCH3), 32.8 (CH2), 29.8 (CH2), 28.0 (CH3), 27.0 (CH3), 24.4 (CH2), 22.5 (CH2) and 14.0 (CH3) ppm. FTIR (film): 1717 (CO) cm−1. MS (ESI) m/z = 373 (MNa+). HRMS calcd for C20H30O5Na (MNa+): 373.1985; found, 373.1982.
:80) diethyl ether/hexane. Yield = 34% (89 mg). Colorless oil. [α]20D = +93.8° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.64 (m, 1H, H2), 5.91 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.44 (dd, J = 1.4 and 17.3 Hz, 1H, CHCHH), 5.18 (dd, J = 1.4 and 10.7 Hz, 1H, CHCHH), 5.01 (br s, 1H, CCHH), 4.97 (br s, 1H, CCHH), 4.07 (m, 1H, H5), 4.04–4.02 (m, 2H, H4 + OCHH), 3.95 (br d, J = 12.5 Hz, 1H, OCHH), 3.76 (s, 3H, OCH3), 2.65 (dd, J = 2.5 and 18.0 Hz, 1H, CHH-6), 2.44 (ddd, J = 2.8, 3.8 and 18.0 Hz, 1H, CHH-6), 1.90 (d, J = 6.8 Hz, 2H, CH2CH(CH2)2), 1.42 (s, 3H, CH3), 1.34 (s, 3H, CH3), 0.84–0.70 (m, 1H, CH2CH(CH2)2), 0.50–0.44 (m, 2H, CH2CHCH2CH2) and 0.08–0.03 (m, 2H, CH2CHCH2CH2) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.6 (C), 146.1 (C), 138.0 (CH), 137.8 (CH), 126.4 (C), 115.2 (CH2), 111.6 (CH2), 109.2 (C), 80.3 (C), 77.5 (CH), 73.3 (CH), 72.6 (CH2), 52.1 (OCH3), 38.2 (CH2), 28.1 (CH3), 27.1 (CH3), 24.4 (CH2), 9.3 (CH), 4.8 (CH2) and 4.7 (CH2) ppm. FTIR (film): 1717 (CO) cm−1. MS (ESI) m/z = 371 (MNa+). HRMS calcd for C20H28O5Na (MNa+): 371.1829; found, 371.1825.
:80) diethyl ether/hexane. Yield = 54% (190 mg). Colorless oil. [α]20D = +85.2° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.63 (t, J = 1.2 Hz, 1H, H2), 5.89 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.43 (dd, J = 1.4 and 17.3 Hz, 1H, CHCHH), 5.16 (dd, J = 1.4 and 10.7 Hz, 1H, CHCHH), 5.12 (m, 2H, CCH2), 4.08–3.96 (m, 4H, H4 + H5 + OCH2), 3.91 (br s, 2H, CH2OEt), 3.74 (s, 3H, OCH3), 3.44 (q, J = 7.0 Hz, 2H, OCH2CH3), 2.64 (dd, J = 2.5 and 18.0 Hz, 1H, CHH-6), 2.43 (ddd, J = 2.8, 3.7 and 18.0 Hz, 1H, CHH-6), 1.40 (s, 3H, CH3), 1.32 (s, 3H, CH3) and 1.18 (t, J = 7.0 Hz, 3H, OCH2CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 142.9 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.1 (CH2), 113.9 (CH2), 109.2 (C), 80.2 (C), 77.4 (CH), 73.7 (CH), 71.4 (CH2), 70.3 (CH2), 65.9 (CH2), 52.0 (OCH3), 28.0 (CH3), 27.0 (CH3), 24.5 (CH2) and 15.3 (CH3) ppm. FTIR (film): 1716 (CO) cm−1. MS (ESI) m/z = 375 (MNa+). HRMS calcd for C19H28O6Na (MNa+): 375.1778; found, 375.1777.
:50) diethyl ether/hexane. Yield = 63% (207 mg). Yellow oil. [α]20D = +85.9° (c 0.8, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 7.38–7.28 (m, 5H, 5 × ArH), 6.65 (t, J = 1.3 Hz, 1H, H2), 5.89 (dd, J = 10.7 and 17.3 Hz, 1H, CHCH2), 5.43 (dd, J = 1.4 and 17.3 Hz, 1H, CHCHH), 5.18–5.14 (m, 3H, CHCHH + CCH2), 4.49 (s, 2H, OCH2), 4.12–4.02 (m, 4H, H4 + H5 + CH2OBn), 3.99 (br s, 2H, OCH2Ph), 3.76 (s, 3H, OCH3), 2.65 (dd, J = 2.5 and 18.0 Hz, 1H, CHH-6), 2.45 (ddd, J = 2.8, 3.7 and 18.0 Hz, 1H, CHH-6), 1.42 (s, 3H, CH3) and 1.34 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 142.6 (C), 138.3 (C), 137.9 (CH), 137.7 (CH), 128.5 (2 × CH), 127.8 (2 × CH), 127.8 (CH), 126.3 (C), 115.2 (CH2), 114.3 (CH2), 109.2 (C), 80.2 (C), 77.4 (CH), 73.7 (CH), 72.3 (CH2), 70.9 (CH2), 70.3 (CH2), 52.0 (OCH3), 28.0 (CH3), 27.0 (CH3) and 24.5 (CH2) ppm. FTIR (film): 1709 (CO) cm−1. MS (ESI) m/z = 437 (MNa+). HRMS calcd for C24H30O6Na (MNa+): 437.1935; found, 437.1936.
:75) diethyl ether/hexane. Yield = 43% (72 mg). Colorless oil. [α]20D = +63.2° (c 1.0, CHCl3). 1H NMR (250 MHz, CDCl3) δ: 6.63 (br s, 1H, H2), 5.89 (dd, J = 10.7 and 17.3 Hz, 1H, HCCH2), 5.43 (dd, J = 1.4 and 17.3 Hz, 1H, HCCHH), 5.18–5.14 (m, 2H, HCCHH + CCHH), 5.04 (br s, 1H, CCHH), 4.10 (br s, 2H, OCH2), 4.07–3.94 (m, 4H, H4 + H5 + CH2OTBS), 3.75 (s, 3H, OCH3), 2.63 (dd, J = 1.9 and 18.0 Hz, 1H, CHH-6), 2.42 (dt, J = 3.3 and 18.0 Hz, 1H, CHH-6), 1.40 (s, 3H, CH3), 1.33 (s, 3H, CH3), 0.89 (s, 9H, C(CH3)3) and 0.04 (s, 6H, 2 × CH3) ppm. 13C NMR (63 MHz, CDCl3) δ: 167.5 (C), 145.0 (C), 137.9 (CH), 137.7 (CH), 126.3 (C), 115.2 (CH2), 112.0 (CH2), 109.2 (C), 80.2 (C), 77.3 (CH), 73.3 (CH), 70.2 (CH2), 63.9 (CH2), 52.1 (OCH3), 28.0 (CH3), 27.0 (CH3), 26.0 (C(CH3)3), 24.4 (CH2), 18.5 (C(CH3)3) and −5.3 (2 × CH3) ppm. FTIR (film): 1716 (CO) cm−1. MS (ESI) m/z = 461 (MNa+). HRMS calcd for C23H38O6SiNa (MNa+): 461.2330; found, 461.2330.
General procedure for ring-closing metathesis of compounds 6a–g and 6i
A solution of compounds 6a–g and 6i (1 mmol) and 2nd generation Grubbs’ catalyst (0.02 mmol) in dry toluene (20 mM), under an inert atmosphere, was heated at 90 °C for 24–48 h. After cooling to room temperature, the mixture was filtered over Celite® and the residue was washed with diethyl ether. The filtrate and the washings were concentrated under reduced pressure and purified by flash chromatography to yield the bicycles 24a–g and 24i.:50) diethyl ether/hexane. Yield = 63% (57 mg). It was also recovered 18 mg of starting material. Corrected yield = 77%. Colorless oil. [α]20D = +11.4° (c 1.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.70 (q, J = 1.7 Hz, 1H, H7), 5.68 (t, J = 1.7 Hz, 1H, H5), 4.50 (m, 1H, H1), 4.35 (dd, J = 5.0 and 1.7 Hz, 1H, H10), 4.28 (br d, J = 16.8 Hz, 1H, OCHH), 3.97 (d, J = 16.8 Hz, 1H, OCHH), 3.72 (s, 3H, OCH3), 2.52 (m, 2H, CH2-9), 1.66 (s, 3H, CH3), 1.39 (s, 3H, CH3) and 1.31 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.6 (C), 140.0 (C), 138.0 (CH), 125.2 (C), 123.9 (CH), 110.1 (C), 79.8 (C), 75.0 (CH), 70.3 (CH), 68.0 (OCH2), 51.9 (OCH3), 28.1 (CH3), 27.3 (CH3), 27.3 (CH2) and 21.9 (CH3) ppm. FTIR (film): 1798 (CO) cm−1. MS (ESI) m/z = 303 (MNa+). HRMS calcd for C15H20O5Na (MNa+): 303.1203; found, 303.1202.
:70) diethyl ether/hexane. Reaction time = 48 h. Yield = 47% (82 mg). It was also recovered 95 mg of starting material. Corrected yield = 97%. Colorless oil. [α]20D = +30.8° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.67 (m, 1H, H7), 5.63 (m, 1H, H5), 4.46 (m, 1H, H1), 4.35–4.26 (m, 2H, H10 + OCHH-3), 3.99 (d, J = 16.6 Hz, 1H, OCHH-3), 3.70 (m, 3H, OCH3), 2.51 (m, 2H, CH2-9), 1.93 (q, J = 7.4 Hz, 2H, CH2CH3), 1.37 (s, 3H, CH3), 1.30 (s, 3H, CH3) and 0.98 (t, J = 7.3 Hz, 3H, CH2CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 145.4 (C), 138.0 (CH), 125.0 (C), 122.1 (CH), 110.0 (C), 80.0 (C), 75.0 (CH), 70.2 (CH), 67.3 (CH2), 51.9 (OCH3), 28.6 (CH2), 28.1 (CH3), 27.3 (CH3), 27.2 (CH2) and 12.0 (CH3) ppm. FTIR (film): 1712 (CO) cm−1. MS (ESI) m/z = 317 (MNa+). HRMS calcd for C16H22O5Na (MNa+): 317.1359; found, 317.1359.
:80) diethyl ether/hexane. Reaction time = 48 h. Yield = 63% (94 mg). It was also recovered 43 mg of starting material. Corrected yield = 89%. Colorless oil. [α]20D = +32.0° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.69 (m, 1H, H7), 5.67 (m, 1H, H5), 4.48 (m, 1H, H1), 4.36 (dd, J = 1.6 and 6.6 Hz, 1H, H10), 4.29 (dd, J = 1.9 and 16.7 Hz, 1H, OCHH-3), 4.01 (d, J = 16.7 Hz, 1H, OCHH-3), 3.73 (m, 3H, OCH3), 2.53 (m, 2H, CH2-9), 1.92 (t, J = 7.5 Hz, 2H, CH2CH2CH3), 1.44–1.36 (m, 5H, CH2CH2CH3 + CH3), 1.33 (s, 3H, CH3) and 0.88 (t, J = 7.3 Hz, 3H, (CH2)2CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 143.8 (C), 137.9 (CH), 125.0 (C), 123.3 (CH), 110.0 (C), 79.9 (C), 74.9 (CH), 70.2 (CH), 67.3 (CH2), 51.9 (OCH3), 38.0 (CH2), 28.1 (CH3), 27.2 (CH3), 27.2 (CH2), 20.8 (CH2) and 13.8 (CH3) ppm. FTIR (film): 1717 (CO) cm−1. MS (ESI) m/z = 331 (MNa+). HRMS calcd for C17H24O5Na (MNa+): 331.1516; found, 331.1513.
:85) diethyl ether/hexane. Reaction time = 24 h. Yield = 54% (54 mg). Colorless oil. [α]20D = 37.5° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.68 (m, 1H, H7), 5.65 (br s, 1H, H5), 4.47 (m, 1H, H1), 4.34 (dd, J = 1.7 and 5.0 Hz, 1H, H10), 4.29 (dd, J = 2.2 and 16.7 Hz, 1H, OCHH-3), 4.00 (d, J = 16.7 Hz, 1H, OCHH-3), 3.72 (m, 3H, OCH3), 2.52 (m, 2H, CH2-9), 1.92 (t, J = 6.9 Hz, 2H, CH2(CH2)2CH3), 1.38 (s, 3H, CH3), 1.36–1.21 (m, 4H, CH2(CH2)2CH3), 1.31 (s, 3H, CH3) and 0.87 (t, J = 7.1 Hz, 3H, (CH2)3CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.6 (C), 144.1 (C), 138.0 (CH), 125.0 (C), 123.1 (CH), 110.1 (C), 79.9 (C), 75.0 (CH), 70.2 (CH), 67.4 (CH2), 51.9 (OCH3), 35.7 (CH2), 29.8 (CH2), 28.1 (CH3), 27.3 (CH3), 27.2 (CH2), 22.5 (CH2) and 14.0 (CH3) ppm. FTIR (film): 1717 (CO) cm−1. MS (ESI) m/z = 345 (MNa+). HRMS calcd for C18H26O5Na (MNa+): 345.1672; found, 345.1670.
:80) diethyl ether/hexane. Reaction time = 48 h. Yield = 45% (43 mg). It was also recovered 59 mg of starting material. Corrected yield = quant. Colorless oil. [α]20D = +30.5° (c 1.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.69 (m, 1H, H7), 5.82 (m, 1H, H5), 4.48 (m, 1H, H1), 4.38–4.30 (m, 2H, H10 + OCHH-3), 4.05 (d, J = 16.7 Hz, 1H, OCHH-3), 3.72 (m, 3H, OCH3), 2.52 (m, 2H, CH2-9), 1.90 (dd, J = 6.4 and 15.8 Hz, 1H, CHH), 1.77 (dd, J = 7.0 and 15.9 Hz, 1H, CHH), 1.40 (s, 3H, CH3), 1.32 (s, 3H, CH3), 0.71 (m, 1H, CH2CH(CH2)2), 0.50–0.45 (m, 2H, CHCH2CH2) and 0.06–0.03 (m, 2H, CHCH2CH2) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.6 (C), 144.0 (C), 138.1 (CH), 125.1 (C), 123.2 (CH), 110.1 (C), 80.0 (C), 75.0 (CH), 70.3 (CH), 67.4 (CH2), 52.0 (OCH3), 40.5 (CH2), 28.1 (CH3), 27.3 (CH3), 27.3 (CH3), 9.0 (CH), 4.8 (CH2) and 4.7 (CH2) ppm. FTIR (film): 1718 (CO) cm−1. MS (ESI) m/z = 343 (MNa+). HRMS calcd for C18H24O5Na (MNa+): 343.1516; found, 343.1524.
:80) diethyl ether/hexane. Yield = 16% (36 mg). It was also recovered 156 mg of starting material. Corrected yield = 78%. Yellow oil. [α]20D = +36.9° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.68 (m, 1H, H7), 5.95 (m, 1H, H5), 4.51 (m, 1H, H1), 4.40 (dd, J = 1.4 and 4.8 Hz, 1H, H10), 4.32 (dd, J = 1.5 and 16.7 Hz, 1H, OCHH-3), 4.16 (d, J = 16.7 Hz, 1H, OCHH-3), 3.84 (sa, 2H, CH2OEt), 3.74 (s, 3H, OCH3), 3.43 (q, J = 7.0 Hz, 1H, OCH2CH3), 2.55 (m, 2H, CH2-9), 1.40 (s, 3H, CH3), 1.33 (s, 3H, CH3) and 1.18 (t, J = 7.0 Hz, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 140.6 (C), 137.4 (CH), 125.9 (CH), 125.6 (C), 110.3 (C), 79.6 (C), 74.8 (CH), 73.1 (CH2), 70.3 (CH), 66.0 (CH2), 65.0 (CH2), 52.0 (OCH3), 28.1 (CH3), 27.3 (CH3), 27.2 (CH2) and 15.2 (CH3) ppm. FTIR (film): 1695 (CO) cm−1. MS (ESI) m/z = 347 (MNa+). HRMS calcd for C17H24O6Na (MNa+): 347.1465; found, 347.1466.
Preparation of 24f from 24i
A solution of the alcohol 24i (34 mg, 0.10 mmol) in dry DMF (0.2 mL), at 0 °C and under argon, was treated with NaH (6.6 mg, 0.17 mmol, ca. 60% w/w in mineral oil). After 30 min stirring, bromoethane (20 μL, 0.17 mmol) was added, the ice bath was removed and the reaction mixture was stirred for 3 h at room temperature. The reaction mixture was diluted with a mixture of (4:1) water/ethyl acetate, the organic layer was separated and the aqueous layer was extracted with ethyl acetate keeping the same proportion. The combined organic extracts were dried (anh. Na2SO4), filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography, eluting with (50:50) ethyl acetate/hexane, to give compound 24f (12 mg, 31%). It was also recovered 4 mg of starting material. Corrected yield = 57%.
:80) diethyl ether/hexane. Yield = 26% (53 mg). It was also recovered 138 mg of starting material. Corrected yield = 94%. Yellow oil. [α]20D = +22.3° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 7.37–7.26 (m, 5H, 5 × ArH), 6.69 (m, 1H, H7), 5.98 (br s, 1H, H5), 4.51 (m, 1H, H1), 4.46 (s, 2H, CH2Ph), 4.39 (dd, J = 1.5 and 4.8 Hz, 1H, H10), 4.33 (dd, J = 1.4 and 16.8 Hz, 1H, OCHH-3), 4.18 (d, J = 16.8 Hz, 1H, OCHH-3), 3.89 (sa, 2H, CH2OBn), 3.74 (s, 3H, OCH3), 2.55 (m, 2H, CH2-9), 1.41 (s, 3H, CH3) and 1.34 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 140.4 (C), 137.9 (C), 137.3 (CH), 128.6 (2 × CH), 127.9 (3 × CH), 126.5 (CH), 125.7 (C), 110.3 (C), 79.6 (C), 74.8 (CH), 72.6 (CH2), 72.5 (CH2), 70.3 (CH), 65.1 (CH2), 52.0 (OCH3), 28.1 (CH3), 27.3 (CH3) and 27.2 (CH2) ppm. FTIR (film): 1716 (CO) cm−1. MS (ESI) m/z = 409 (MNa+). HRMS calcd for C22H26O6Na (MNa+): 409.1622; found, 409.1619.
Preparation of 24g from 24i
A solution of the alcohol 24i (31 mg, 0.10 mmol) in dry THF (0.3 mL), at 0 °C and under argon, was treated with NaH (6 mg, 0.15 mmol, ca. 60% w/w in mineral oil). After 30 min stirring, benzyl bromide (20 μL, 0.15 mmol) was added, the ice bath was removed and the reaction mixture was stirred for 3 h at room temperature. Saturated NH4Cl was added, the organic solvent was removed under reduced pressure and the aqueous solution was extracted with ethyl acetate (×3). The combined organic extracts were dried (anh. Na2SO4), filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography, eluting with (25:75) ethyl acetate/hexane, to give compound 24g (25 mg, 64%).
:80) diethyl ether/hexane. Yield = 42% (52 mg). Colorless oil. [α]20D = +33.9° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.68 (m, 1H, H7), 5.92 (m, 1H, H5), 4.51 (m, 1H, H1), 4.39 (dd, J = 1.6 and 4.8 Hz, 1H, H10), 4.28 (dd, J = 1.4 and 16.7 Hz, 1H, OCHH-3), 4.08 (d, J = 16.7 Hz, 1H, OCHH-3), 4.01 (dd, J = 1.3 and 4.6 Hz, 2H, CH2), 3.73 (s, 3H, OCH3), 2.54 (m, 2H, CH2-9), 1.40 (s, 3H, CH3), 1.33 (s, 3H, CH3), 0.89 (s, 9H, C(CH3)3), 0.05 (s, 3H, CH3) and 0.05 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.5 (C), 142.7 (C), 137.6 (CH), 125.4 (C), 123.3 (CH), 110.2 (C), 79.8 (C), 74.9 (CH), 70.3 (CH), 65.3 (CH2), 64.7 (CH2), 52.0 (OCH3), 28.1 (CH3), 27.2 (CH3), 27.2 (CH2), 26.0 (C(CH3)3), 18.5 (C(CH3)3), −5.2 (CH3) and −5.2 (CH3) ppm. FTIR (film): 1716 (CO) cm−1. MS (ESI) m/z = 433 (MNa+). HRMS calcd for C21H34O6SiNa (MNa+): 433.2017; found, 433.2013.
:50) diethyl ether/hexane, to give the alcohol 24h (31 mg, 81%) as a colorless oil. [α]20D = +37.1° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.68 (d, J = 1.0 Hz, 1H, H7), 5.97 (br s, 1H, H5), 4.52 (m, 1H, H1), 4.40–4.32 (m, 2H, H10 + OCHH-3), 4.18 (d, J = 16.7 Hz, 1H, OCHH), 4.02 (t, J = 4.3 Hz, 2H, CH2), 3.74 (s, 3H, OCH3), 2.55 (m, 2H, CH2-9), 1.40 (s, 3H, CH3) and 1.34 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.3 (C), 142.9 (C), 137.2 (CH), 125.5 (C), 124.4 (CH), 110.2 (C), 79.5 (C), 74.6 (CH), 70.2 (CH), 65.0 (CH2), 64.5 (CH2), 51.9 (CH3), 28.0 (CH3), 27.1 (CH3) and 27.0 (CH2) ppm. FTIR (film): 3441 (OH) and 1709 (CO) cm−1. MS (ESI) m/z = 319 (MNa+). HRMS calcd for C15H20O6Na (MNa+): 319.1152; found, 319.1152.
General procedure for the acetal deprotection in 24a–h
A solution of the acetals 24a–h (1 mmol) in methanol (7 mL) and aqueous HCl (8.6 mL, 6 M) was heated at 60 °C for 6 h. The mixture was cooled and concentrated under reduced pressure. The residue was purified by flash chromatography to yield the diols 25a–h.:50) ethyl acetate/hexane. Yield = 88% (30 mg). Colorless oil. [α]20D = −86.6° (c 3.0, CH3OH). 1H NMR (300 MHz, CD3OD) δ: 6.64 (m, 1H, H7), 5.46 (m, 1H, H5), 4.25 (m, 2H, H1 + OCHH), 4.12 (dd, J = 4.9 and 1.6 Hz, 1H, H10), 3.85 (d, J = 16.5 Hz, 1H, OCHH), 2.59 (dt, J = 17.9 and 2.9 Hz, 1H, CHH-9), 2.28 (dd, J = 17.9 and 2.3 Hz, 1H, CHH-9) and 1.68 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CD3OD) δ: 168.3 (C), 144.0 (CH), 140.2 (C), 139.9 (CH), 129.4 (C), 128.7 (CH), 75.4 (CH), 72.6 (C), 70.6 (CH), 67.5 (CH2), 28.3 (CH2) and 22.2 (CH3) ppm. FTIR (ATR): 3416 (OH) and 1698 (CO) cm−1. MS (ESI) m/z = 263 (MNa+). HRMS calcd for C12H16O5Na (MNa+): 263.0890; found, 263.0881.
:20) diethyl ether/hexane. Yield = 80% (87 mg). Colorless oil. [α]20D = −48.1° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.69 (br s, 1H, H7), 5.48 (d, J = 1.4 Hz, 1H, H5), 4.32 (m, 1H, H1), 4.20–4.14 (m, 2H, H10 + OCHH-3), 3.93 (d, J = 16.4 Hz, 1H, OCHH-3), 3.72 (m, 3H, OCH3), 3.07 (br s, 2H, 2 × OH), 2.61 (dt, J = 2.9 and 18.3 Hz, 1H, CHH-9), 2.40 (dd, J = 1.7 and 18.2 Hz, 1H, CHH-9), 1.93 (t, J = 7.4 Hz, 2H, CH2CH2CH3), 1.38 (m, 2H, CH2CH2CH3) and 0.87 (t, J = 7.3 Hz, 3H, (CH2)2CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.8 (C), 143.6 (C), 138.8 (CH), 127.3 (C), 127.0 (CH), 73.3 (CH), 72.0 (C), 69.7 (CH), 66.2 (CH2), 52.0 (OCH3), 38.6 (CH2), 27.2 (CH2), 21.0 (CH2) and 13.8 (CH3) ppm. FTIR (film): 3412 (OH) and 1700 (CO) cm−1. MS (ESI) m/z = 291 (MNa+). HRMS calcd for C14H20O5Na (MNa+): 291.1203; found, 291.1202.
:10) diethyl ether/hexane. Yield = 89% (95 mg). Colorless oil. [α]20D = −42.3° (c 1.0, CH3OH). 1H NMR (300 MHz, CD3OD) δ: 6.68 (br s, 1H, H7), 5.48 (br s, 1H, H5), 4.31–4.26 (m, 2H, H1 + OCHH-3), 4.16 (br d, J = 4.8 Hz, 1H, H10), 3.94 (d, J = 16.3 Hz, 1H, OCHH-3), 3.77 (s, 3H, OCH3), 2.63 (td, J = 2.8 and 18.0 Hz, 1H, CHH-9), 2.32 (br d, J = 18.5 Hz, 1H, CHH-9), 2.02 (t, J = 6.5 Hz, 2H, CH2(CH2)2CH3), 1.44–1.34 (m, 4H, CH2(CH2)2CH3) and 0.95 (t, J = 6.8 Hz, 3H, (CH2)3CH3) ppm. 13C NMR (75 MHz, CD3OD) δ: 169.2 (C), 144.1 (C), 140.9 (CH), 128.9 (CH), 127.8 (C), 75.1 (CH), 72.5 (C), 70.5 (CH), 66.8 (CH2), 52.3 (OCH3), 37.2 (CH2), 31.1 (CH2), 28.2 (CH2), 23.3 (CH2) and 14.3 (CH3) ppm. FTIR (film): 3419 (OH) and 1710 (CO) cm−1. MS (ESI) m/z = 305 (MNa+). HRMS calcd for C15H22O5Na (MNa+): 305.1359; found, 305.1357.
:10) diethyl ether/hexane. Yield = 76% (107 mg). White solid. [α]20D = −44.4° (c 1.0, CHCl3). Mp: 128.2–128.7 °C. 1H NMR (300 MHz, CDCl3) δ: 6.74 (m, 1H, H7), 5.66 (m, 1H, H5), 4.38 (m, 1H, H1), 4.28–4.22 (m, 2H, OCHH-3 + H10), 4.01 (d, J = 16.4 Hz, 1H, OCHH-3), 3.74 (s, 3H, OCH3), 2.65 (dt, J = 3.0 and 18.3 Hz, 1H, CHH-9), 2.45 (dd, J = 2.1 and 18.3 Hz, 1H, CHH-9), 1.88 (m, 2H, CH2), 0.78–0.65 (m, 1H, CH2CH(CH2)2), 0.52–0.46 (m, 2H, CHCH2CH2), 0.07 (dd, J = 1.3 and 5.0 Hz, 1H, CHCH2CHH) and 0.04 (dd, J = 1.4 and 4.7 Hz, 1H, CHCH2CHH) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.8 (C), 143.7 (C), 138.8 (CH), 127.3 (C), 126.8 (CH), 73.3 (CH), 72.0 (C), 69.7 (CH), 66.3 (CH2), 52.0 (OCH3), 41.0 (CH2), 27.2 (CH2), 9.1 (CH), 4.8 (CH2) and 4.7 (CH2) ppm. FTIR (film): 3393 (OH) and 1685 (CO) cm−1. MS (ESI) m/z = 303 (MNa+). HRMS calcd for C15H20O5Na (MNa+): 303.1203; found, 303.1200.
:30) ethyl acetate/hexane. Yield = 80% (34 mg). Brown oil. [α]20D = −35.1° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ: 6.70 (br s, 1H, H7), 5.77 (br s, 1H, H5), 4.38 (q, J = 2.3 Hz, 1H, H1), 4.24–4.19 (m, 2H, H10 + OCHH-3), 4.08 (d, J = 16.5 Hz, 1H, OCHH-3), 3.85 (br s, 2H, CH2OEt), 3.74 (s, 3H, OCH3), 3.44 (q, J = 7.0 Hz, 2H, OCH2CH3), 2.65 (dt, J = 2.9 and 18.4 Hz, 1H, CHH-9), 2.45 (br d, J = 17.9 Hz, 1H, CHH-9) and 1.18 (t, J = 7.0 Hz, 3H, OCH2CH3) ppm. 13C NMR (75 MHz, CDCl3) δ: 167.6 (C), 140.2 (C), 138.1 (CH), 129.7 (CH), 127.9 (C), 73.5 (CH), 73.4 (CH2), 71.8 (C), 69.6 (CH), 66.1 (CH2), 64.0 (CH2), 52.1 (OCH3), 27.2 (CH2) and 15.2 (CH3) ppm. FTIR (film): 3406 (OH) and 1709 (CO) cm−1. MS (ESI) m/z = 307 (MNa+). HRMS calcd for C14H20O6Na (MNa+): 307.1152; found, 307.1153.
General procedure for the ester hydrolysis in 25a–h
A solution of the esters 25a–h (1 mmol) in THF (10 mL) was treated at room temperature with an aqueous solution of LiOH (1.2 mL, 2.5 M). After stirring for 1 h, water was added and THF was removed under reduced pressure. The aqueous layer was washed with ethyl acetate (×3) and then treated with Amberlite IR-120 (H+) until pH 6. The resin was filtered off and washed with MilliQ water. The filtrate and the washings were lyophilized to give acids 4.C14H17O5 (M − H): 265.1081; found, 265.1079.
:95 → 10:90) CH3CN/H2O; (2) 5–20 min (10:90 → 20:80) CH3CN/H2O], at a flow rate of 3.5 mL min−1.
Docking studies
They were carried out using the GOLD 5.2.2 program and the enzyme coordinates found in the crystal structures of Hp-SK in complex with shikimate-3-phosphate and ADP (PDB entry 3MUF,15 2.3 Å) and of Mt-SK in complex with 2 and ADP (PDB entry 4BQS,13 2.15 Å). Ligand geometries were minimized using the AM1 Hamiltonian as implemented in the program Gaussian 0920 and used as MOL2 files. Each ligand was docked in 25 independent genetic algorithm (GA) runs, and for each of these a maximum number of 100000 GA operations were performed on a single population of 50 individuals. Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95, and 10, respectively), as well as the hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. The position of shikimate-3-phosphate and compound 2 present in the aforementioned PDB files were used to define the active-site and the radius was set to 8 Å. All crystallographic water molecules and the aforementioned ligands were removed for docking. The “flip ring corners” flag was switched on, while all the other flags were off. The GOLD scoring function was used to rank the ligands in order to fitness. The molecular graphics program PyMOL was employed for visualization and depicting ligand/protein structures.21
Molecular dynamics simulation studies
Abbreviations
| SK | Shikimate kinase |
| Hp-SK | Shikimate kinase from Helicobacter pylori |
| Mt-SK | Shikimate kinase from Mycobacterium tuberculosis |
| MD | Molecular dynamics |
| PDB | Protein data bank |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competiveness (SAF2016-75638-R), the Xunta de Galicia [Centro singular de investigación de Galicia accreditation 2016–2019 (ED431G/09) and ED431B 2018/04], and the European Union (European Regional Development Fund – ERDF) is gratefully acknowledged. MP and EL thank the Xunta de Galicia for their respective predoctoral and postdoctoral fellowships. We are grateful to the Centro de Supercomputación de Galicia (CESGA) for use of computational facilities.Notes and references
- C. E. Chang, W. Chen and M. K. Gilson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1534–1539 CrossRef CAS.
- M. H. Hao, O. Haq and I. Muegge, J. Chem. Inf. Model., 2007, 47, 2242–2252 CrossRef CAS PubMed.
- M. Sitzmann, I. E. Weidlich, I. V. Filippov, C. Liao, M. L. Peach, W.-D. Ihlenfeldt, R. G. Karki, Y. V. Borodina, R. E. Cachau and M. C. Nicklaus, J. Chem. Inf. Model., 2012, 52, 739–756 CrossRef CAS PubMed.
- J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2006, 49, 5880–5884 CrossRef CAS PubMed.
- Z. Fang, Y. Song, P. Zhan, Q. Zhang and X. Liu, Future Med. Chem., 2014, 6, 885–901 CrossRef CAS PubMed.
- R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359–1472 CrossRef CAS.
- E. Perola and P. S. Charifson, J. Med. Chem., 2004, 47, 2499–2510 CrossRef CAS PubMed.
- G. Klebe, F. Dullweber and H.-J. Böhm, Thermodynamic models of drug-receptor interactions: a general introduction, in Drug-Receptor Thermodynamics: Introduction and Applications, John Wiley & Sons, Chichester, U.K., 2001, pp. 83–104 Search PubMed.
- J. Boström and A. Grant, Molecular drug properties: measurement and prediction, in Exploiting Ligand Conformations in Drug Design, Wiley-VCH, Weinheim, Germany, 2007, vol. 37, pp. 183–205 Search PubMed.
- R. K. Tiwari and K. Parang, Curr. Pharm. Des., 2012, 18, 2852–2866 CrossRef CAS PubMed.
- Y. Wang, A. Kirschner, A.-K. Fabian, R. Gopalakrishnan, C. Kress, B. Hoogeland, U. Koch, C. Kozany, A. Bracher and F. Hausch, J. Med. Chem., 2013, 56, 3922–3935 CrossRef CAS PubMed.
- (a) A. T. Namanja, X. J. Wang, B. Xu, A. Y. Mercedes-Camacho, B. D. Wilson, K. A. Wilson, F. A. Etzkorn and J. W. Peng, J. Am. Chem. Soc., 2010, 132, 5607–5609 CrossRef CAS PubMed; (b) D. Quaglio, G. Zappia, E. De Paolis, S. Balducci, B. Botta and F. Ghirga, Org. Chem. Front., 2018, 5, 3022–3055 RSC.
- B. Blanco, V. Prado, E. Lence, J. M. Otero, C. García-Doval, M. J. van Raaij, A. L. Llamas-Saiz, H. Lamb, A. R. Hawkins and C. González-Bello, J. Am. Chem. Soc., 2013, 135, 12366–12376 CrossRef CAS PubMed.
- http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ .
- W. C. Cheng, Y. F. Chen, H. J. Wang, K. C. Hsu, S. C. Lin, T. J. Chen, J. M. Yang and W. C. Wang, PLoS One, 2012, 7, e33481 CrossRef CAS PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 2015, University of California, San Francisco, 2015 Search PubMed.
- Y. Dai, F. Wu, Z. Zang, H. You and H. Gong, Chem. – Eur. J., 2012, 18, 808–812 CrossRef CAS PubMed.
- V. Prado, E. Lence, J. A. Vallejo, A. Beceiro, P. Thompson, A. R. Hawkins and C. González-Bello, Chem. – Eur. J., 2016, 22, 2758–2768 CrossRef CAS PubMed.
- V. Prado, E. Lence, P. Thompson, A. R. Hawkins and C. González-Bello, Chem. – Eur. J., 2016, 22, 17988–18000 CrossRef CAS PubMed.
- M. J. Frisch, et al., Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific LLC, Palo Alto, CA, USA, 2008, http://www.pymol.org/ Search PubMed.
- (a) W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS; (b) http://upjv.q4md-forcefieldtools.org/RED/ (accessed March 1, 2019).
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, O. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- (a) J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS; (b) J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS.
- (a) J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, Nucleic Acids Res., 2005, 33(Web Server issue), W368–W371 CrossRef CAS PubMed; (b) http://biophysics.cs.vt.edu/H++ (accessed March 1, 2019).
- (a) J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed; (b) J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- D. A. Case, et al., AMBER 2015, University of California, San Francisco, 2015 Search PubMed.
- Mg2+ parameters used with the AMBER force field were downloaded from http:/www.pharmacy.manchester.ac.uk/bryce/amber/ CrossRef PubMed(accessed March 1, 2019). O. Allnér, L. Nilsson and A. Villa, J. Chem. Theory Comput., 2012, 8, 1493–1502 CrossRef PubMed.
- ATP parameters used with the AMBER force field were downloaded from http://www.pharmacy.manchester.ac.uk/bryce/amber/ (accessed March 1, 2019). K. L. Meagher, L. T. Redman and H. A. Carlson, J. Comput. Chem., 2003, 24, 1016–1025 CrossRef CAS PubMed.
- J. Aqvist, J. Phys. Chem., 1990, 94, 8021–8024 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar and J. D. Madura, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- (a) A. W. Goetz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef; (b) R. Salomon-Ferrer, A. W. Goetz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed; (c) S. Le Grand, A. W. Goetz and R. C. Walker, Comput. Phys. Commun., 2013, 184, 374–380 CrossRef CAS.
- T. A. Darden, D. York and L. G. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the experimental procedures for the synthesis of carbonates 8, inhibition assays and extra figures. See DOI: 10.1039/c9qo00562e |
| This journal is © the Partner Organisations 2019 |