
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxylammonium derivatives for selective active-site lysine modification in the anti-virulence bacterial target DHQ1 enzyme†
María
Maneiro
 a,
Emilio
Lence
a,
Marta
Sanz-Gaitero
b,
José M.
Otero
a,
Mark J.
van Raaij
b,
Paul
Thompson
c,
Alastair R.
Hawkins
c and
Concepción
González-Bello
*a
a,
Emilio
Lence
a,
Marta
Sanz-Gaitero
b,
José M.
Otero
a,
Mark J.
van Raaij
b,
Paul
Thompson
c,
Alastair R.
Hawkins
c and
Concepción
González-Bello
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, Jenaro de la Fuente s/n, 15782 Santiago de Compostela, Spain. E-mail: concepcion.gonzalez.bello@usc.es
bDepartamento de Estructura de Macromoléculas, Centro Nacional de Biotecnología (CSIC), Campus Cantoblanco, 28049 Madrid, Spain
cInstitute of Cell and Molecular Biosciences, Medical School, University of Newcastle upon Tyne, Newcastle upon Tyne NE2 4HH, UK
First published on 23rd July 2019
Abstract
Targeted irreversible inhibitors bearing electrophiles that become activated towards covalent bond formation upon binding to a specific protein/enzyme is an emerging area in drug discovery. Targeting lysine residues is challenging due to the intrinsically low reactivity of the amino group at physiological pH. Herein we report the first example of a hydroxylammonium derivative that causes a specific covalent modification of an active-site and a sterically inaccessible lysine residue of an enzyme. The described ligands, compounds 1–3, were rationally designed to be activated towards covalent bond formation upon binding to the type I dehydroquinase (DHQ1) enzyme for the development of new anti-virulence agents to combat the widespread resistance to antibiotics. Evidence in atomic detail for the covalent modifications caused by the ligands to the catalytic Lys170 by the formation of a stable secondary amine is provided by the resolution at 1.08–1.25 Å of the crystal structures of DHQ1 from Salmonella typhi enzyme adducts. In addition, the first crystal structure of the addition intermediate adduct at 1.4 Å of a Schiff base formation reaction by using an analog of the natural substrate, compound 4, is also reported. Molecular dynamics simulation studies on non-covalent enzyme/ligand complexes and a two-dimensional QM/MM umbrella sampling simulation study suggested that a direct displacement by Lys170 with the release of NH2OH would be feasible. These studies might open up new opportunities for the development of novel lysine-targeted irreversible inhibitors bearing a methylhydroxylammonium moiety as a latent electrophile.
Introduction
Targeting infectious diseases and cancer with small molecules that covalently modify the target is an emerging area in drug discovery.1–9 The more detailed knowledge available today on the real risks associated with this type of compound, as well as its enormous advantages (efficacy, selectivity, and lower susceptibility to resistance mechanisms), has led to a dramatic increase in their presence in anti-infective and oncology discovery programs in recent years.10 Besides these advantages, the development of this type of ligand represents a huge challenge due to the need to combine reactivity (covalent modification) and selectivity (safety) in a single chemical entity. This indicates the use of ligands bearing weak electrophilic groups which are able to achieve potency without sacrificing selectivity. In recent years, efforts have been devoted to the design of irreversible compounds capable of modulating their reactivity when complementarity with the specific target takes place.8 These are targeted irreversible inhibitors bearing electrophiles which become activated towards covalent bond formation upon binding to a specific protein or enzyme. The intrinsically low chemical reactivity of these ligands makes them particularly desirable for drug development since they would be hidden to non-specific targets and are only activated for the chemical modification reaction upon binding. The majority of these ligands have been designed to modify cysteine residues. Relevant examples include fluoromethylketones, terminal alkynes, acrylamides, and disulfides (Fig. 1A).11–18 Targeting lysine residues appears more challenging because their ε-amino group is usually non-nucleophilic as it is protonated at physiological pH.19 Remarkable examples include aromatic sulfonyl fluorides, vinyl sulfones, aryl fluorosulfates and nitriles (Fig. 1B).20–26 The selective modification of active-site lysine residues is more affordable, because they either have a lower pKa or their nucleophilicity/basicity is modulated by other amino acid residues (Asp and His) in the vicinity, which might also participate in the catalysis. | ||
| Fig. 1 Selected examples of latent electrophiles and targeted ligands. (A and B) Latent electrophiles for modification of cysteine (A) and lysine residues (B). The position that is covalently modified is indicated with an asterisk. (C) Designed quinic acid-based hydroxylammonium compounds 1–3 and substrate analog 4. The chemical modifications of the catalytic Lys170 of DHQ1 from S. typhi identified by X-ray crystallography are also shown. | ||
We became interested in the design of ligands able to cause the covalent modification of the type I dehydroquinase (DHQ1) enzyme. DHQ1 is an aldolase (dehydratase) enzyme that does not have any counterpart in human cells, and has been pinpointed as a promising target in the search for new anti-virulence agents.27 It is believed that DHQ1 may act as a virulence factor in vivo since the deletion of the aroD gene, which encodes DHQ1 from Salmonella typhi and Shigella flexneri, has been proven to afford satisfactory live oral vaccines.28–30 Targeting the bacterial capacity to produce infection (virulence), rather than the most widely used disruption of bacterial survival, is an innovative strategy which is increasingly being explored to combat the worldwide increasing appearance of “superbugs” (multi-drug resistant bacteria), which are resistant to most antibiotics in clinical use. Compounds that target bacterial virulence signalling pathways would create an in vivo scenario similar to vaccination with a live attenuated strain in which the bacteria are eventually cleared by the host immune system. DHQ1 is present in several pathogenic bacteria such as Escherichia coli, Staphylococcus aureus and Salmonella typhi. This enzyme, which is a dimer, has 8 lysine residues in each chain and catalyzes the reversible syn dehydration of water in 3-dehydroquinic acid to form 3-dehydroshikimic acid by a multi-step mechanism which involves the formation of Schiff base species.31
We report here the use of hydroxylammonium derivatives, compounds 1–3, for the specific covalent modification of the catalytic lysine residue of DHQ1 from S. typhi (St-DHQ1) (Fig. 1C). Evidence in atomic detail of the covalent modifications caused by compounds 1–3 to the essential Lys170 by the formation of a stable amine is provided by the resolution of high quality crystal structures of St-DHQ1 (resolution range: 1.08–1.25 Å) chemically modified by 1–3. The reported compounds were also designed to provide non-labile enzyme adducts by being able to avoid the intrinsic dehydration reaction performed by the enzyme. The chemical basis of the latter was inspired by the herein reported Molecular Dynamics (MD) simulation studies on the Michaelis and substrate Schiff base complexes, as well as by the resolution for the first time of the crystal structure of the addition intermediate adduct at 1.4 Å of the substrate Schiff base formation reaction of St-DHQ1 by using an analog of the natural substrate, compound 4.
Results and discussion
Ligand design
It has been proposed that the reaction catalyzed by DHQ1 is initiated by the activation of the C3 carbonyl group of the substrate from the Re face by the essential His143, and subsequent nucleophilic attack of the ε-amino group of the essential Lys170 from the opposite face to afford initially the addition intermediate adduct (Fig. 2A).32 It has been shown previously that epoxide 6 causes the chemical modification of St-DHQ1 by the formation, after dehydration, of a Schiff base with the essential Lys170, adduct II (Fig. 2B).32 This process was evidenced by the resolution of the crystal structure of the St-DHQ1/6 adduct obtained by co-crystallization (PDB entry 4CLM). On the other hand, derivative 5 proved to cause the covalent modification of Lys170 through the initial formation of an amine, adduct I, as revealed by the crystal structure of the enzyme adduct obtained by soaking apo-St-DHQ1 crystals for 30 s (PDB entry 4UIO).33 In solution, this initial adduct I undergoes a dehydration reaction to afford also the imine adduct II (S. typhi).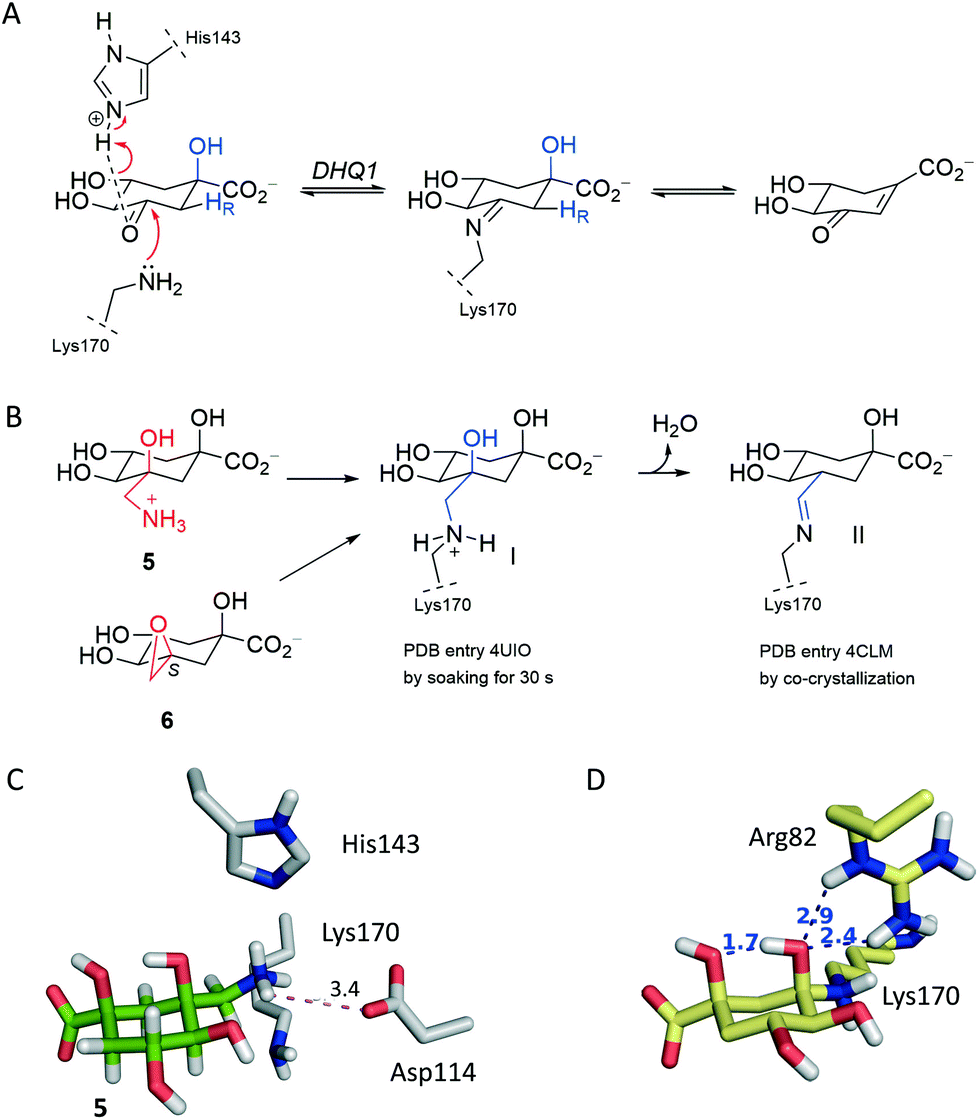 | ||
| Fig. 2 Mechanistic and structural basis for ligand design. (A) Reaction catalyzed by DHQ1. The role of the essential residues Lys170 and His143 in the formation of the substrate-Schiff base in the enzymatic mechanism is shown. (B) Chemical modification of St-DHQ1 by 5 and 6 identified by X-ray crystallography. (C) Arrangement of 5 relative to Asp114. (D) Substrate addition intermediate adduct after 50 ns of dynamic simulation (yellow) obtained by computational studies. | ||
Considering that the formation of an imine group does not represent the best scenario for disabling a target, even taking into account that it proved to be a very stable Schiff base, our efforts were devoted to not only exploring the potential of a methylhydroxylammonium group as a novel latent electrophile, but also avoiding possible dehydration processes by using another type of covalent linkage with the protein. The hydroxylammonium group was chosen because: (i) in MD simulation studies on the St-DHQ1/5 complex, it was observed that the ammonium group in 5 establishes, during a significant part of the simulation, a weak electrostatic interaction (∼3.4 Å) with the conserved Asp114 residue (Fig. 2C); and (ii) the incorporation of an OH group in the leaving group would enhance the latter interaction and therefore would fix the arrangement of the side chain for nucleophilic attack by Lys170 from the opposite site.
Moreover, MD simulation studies carried out with the addition intermediate adduct revealed that the C3 hydroxyl group in the latter adduct remained fixed during the whole simulation (50 ns) with the proton engaged in an intramolecular hydrogen bond with one of the oxygen lone pairs of the C1 hydroxyl group and with one of the oxygen lone pairs accepting a hydrogen bond from the guanidinium group of Arg48 (Fig. 2D and S1†). The C1 hydroxyl group seems to be therefore employed by DHQ1 to achieve the appropriate arrangement of the C3 hydroxyl group for an effective subsequent dehydration reaction to afford the substrate Schiff base. Reasoning that the dehydration reaction would be disfavored by using compounds lacking the tertiary C1 hydroxyl group, compound 4 was synthesized for structural studies that could confirm this hypothesis.
Synthesis of ligands 1–4
Ligand 4 was prepared as outlined in Fig. 3A. Firstly, hydrogenolysis of the protected 3-epi-shikimic acid 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 afforded compound 8 diastereoselectively. It is important to highlight that having the S configuration in the secondary alcohol is crucial to achieve good stereocontrol of the reduction from the Re face of the double bond. The configuration of the new chiral center in 8 was confirmed by the coupling constants in the corresponding 1H NMR spectra (Fig. S2†). Basic hydrolysis of 8 followed by alkylation with benzyl bromide gave ester 10. Oxidation of 10 using Dess–Martin periodinane followed by acid hydrolysis of the acetal group in 11 and final hydrogenolysis of the resulting benzyl ester 12 gave acid 4.
34 afforded compound 8 diastereoselectively. It is important to highlight that having the S configuration in the secondary alcohol is crucial to achieve good stereocontrol of the reduction from the Re face of the double bond. The configuration of the new chiral center in 8 was confirmed by the coupling constants in the corresponding 1H NMR spectra (Fig. S2†). Basic hydrolysis of 8 followed by alkylation with benzyl bromide gave ester 10. Oxidation of 10 using Dess–Martin periodinane followed by acid hydrolysis of the acetal group in 11 and final hydrogenolysis of the resulting benzyl ester 12 gave acid 4.
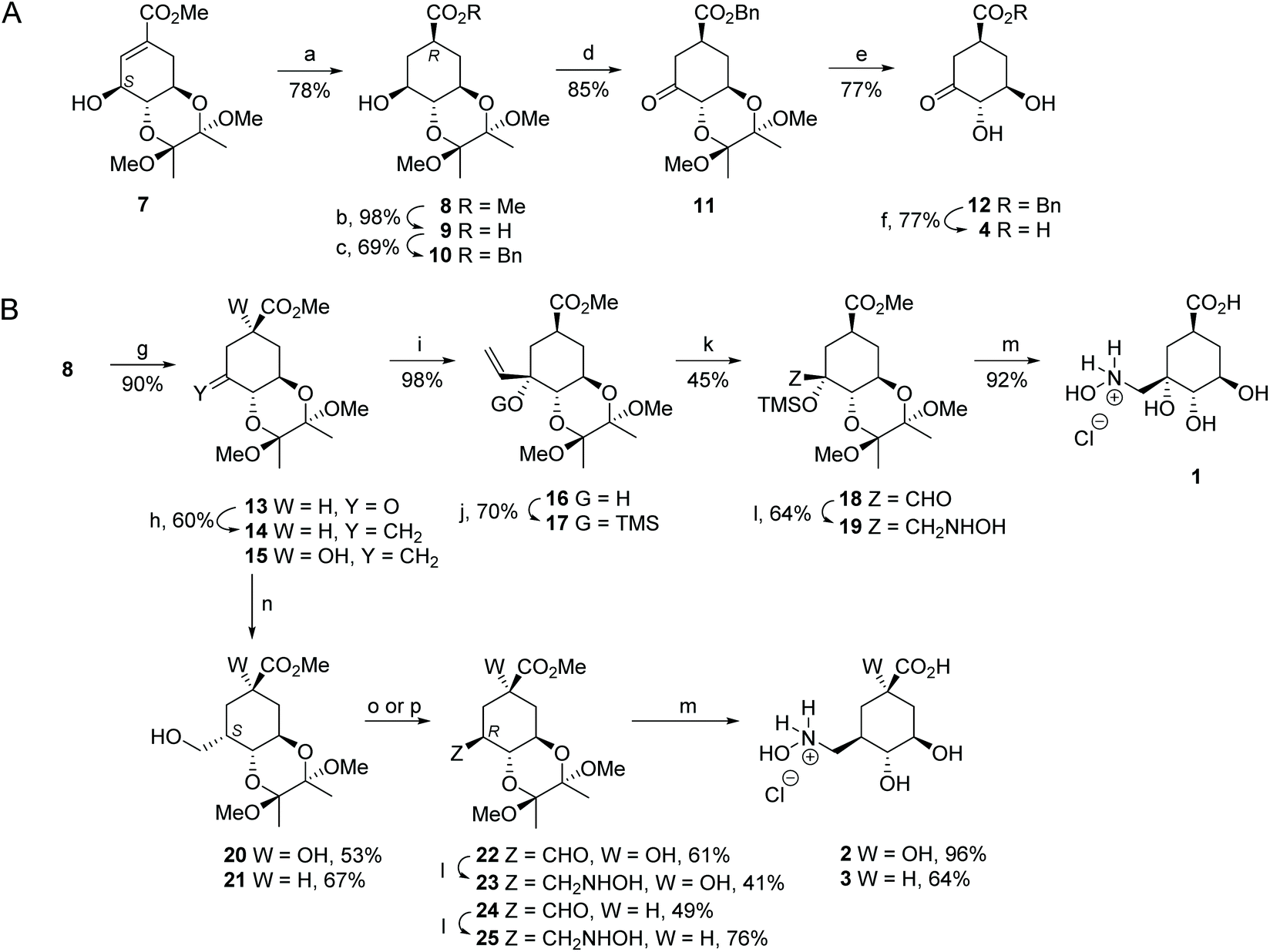 | ||
Fig. 3 Synthesis of compounds 4 (A) and 1–3 (B). Reagents and conditions: (a) H2 (g), Pd/C (10%), MeOH, RT. (b) 1. LiOH, THF, RT; 2. Amberlite IR-120 (H+). (c) 1. E3N, DMAP, Bu4NI, DMF, RT; 2. BnBr, RT. (d) Dess–Martin periodinane, DCM, RT. (e) TFA/H2O (20:1), RT. (f) H2, Pd/C (10%), EtOAc, RT. (g) PDC, 4 Å MS, DCM, RT. (h) Zn dust, CH2I2, TiCl4, THF, 0 °C. (i) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMgBr, THF, −78 °C. (j) TMSCl, (TMS)2NH, Py, RT. (k) 1. O3, DCM, −78 °C. 2. SMe2, −78 °C. (l) 1. NH2OH.HCl, NaOAc, 4 Å MS, MeOH, RT; 2. NaBH3CN, MeOH, RT. (m) HCl (0.3 M), 100 °C. (n) 1. BH3-THF, THF, 0 °C; 2. NaBO3. (o) for 20:1. Dess–Martin periodinane, DCM, RT; 2. Py, MeOH, 60 °C. (p) for 21:1. PDC, 4 Å MS, DCM, RT; 2. Py, MeOH, 60 °C. CHMgBr, THF, −78 °C. (j) TMSCl, (TMS)2NH, Py, RT. (k) 1. O3, DCM, −78 °C. 2. SMe2, −78 °C. (l) 1. NH2OH.HCl, NaOAc, 4 Å MS, MeOH, RT; 2. NaBH3CN, MeOH, RT. (m) HCl (0.3 M), 100 °C. (n) 1. BH3-THF, THF, 0 °C; 2. NaBO3. (o) for 20:1. Dess–Martin periodinane, DCM, RT; 2. Py, MeOH, 60 °C. (p) for 21:1. PDC, 4 Å MS, DCM, RT; 2. Py, MeOH, 60 °C. | ||
Compound 1 was synthesized from aldehyde 18, which was prepared in four steps from alcohol 8 (Fig. 3B). Firstly, PDC-oxidation of 8 followed by nucleophilic addition of vinyl magnesium bromide to the resulting ketone 13 gave diastereoselectively the vinyl derivative 16. The regioselectivity of the reaction was confirmed by NOE experiments. Inversion of the signal of H6 led to an enhancement of the signal for the H1′ vinyl group (3.1%). Having observed that the ozonolysis of alkene 16 afforded complex reaction mixtures and a very low yield of the desired compound, protection of the tertiary alcohol in 16 as a trimethylsilyl ether was carried out. In this way, ozonolysis of the resulting protected alkene 17 and subsequent reductive workup using dimethyl sulfide gave the desired aldehyde 18. Finally, reductive amination of aldehyde 18 with hydroxylamine followed by hydrolysis of methyl ester 19 under acidic conditions with concomitant removal of the protecting groups led to the target compound 1.
Compounds 2 and 3 were prepared from previously reported alkene 1532 and compound 14 using aldehydes 22 and 24 as key intermediates, respectively (Fig. 3B). Alkene 14 was synthesized by methylenation of the ketone 15. Subsequent conversion of the alkenes 15 and 14 to the required aldehydes 22 and 24, respectively, was achieved in two steps. Firstly, hydroboration–oxidation took place from the less hindered Si face of the external double bond to afford compounds 20 and 21 diastereoselectively, both of which have the CH2OH group in the axial disposition. Secondly, oxidation of the primary alcohols in 20 and 21 using Dess–Martin periodinane or PDC with subsequent full epimerization to the thermodynamic equatorial aldehydes by heating in the presence of pyridine gave the desired aldehydes 22 and 24, respectively. The stereochemistry of the new chiral center created in compounds 20–22 and 24 was confirmed by the coupling constants in the corresponding 1H NMR spectra and by NOE experiments (Fig. S2†). Finally, aldehydes 22 and 24 were converted to the target derivatives 2 and 3 in the same way as that for compound 1 from aldehyde 18.
X-ray crystal structures of St-DHQ1/1–4 adducts
 | ||
| Fig. 4 Crystal structure of St-DHQ1 covalently modified by 4. (A) Overall view of the structure of St-DHQ1 covalently modified by 4. (B) Unbiased electron density for the modified ligand 4 (yellow) and its covalent attachment to Lys170 (green) of St-DHQ1 (gray). A maximum-likelihood weighted 2Fo − Fc map contoured at 1σ is shown up to 1.6 Å around the ligand molecule. The final model, including the ligand molecule, is superimposed onto the map. (C) Interactions of the modified ligand 4 with St-DHQ1. Hydrogen bonding and electrostatic interactions between the ligand and St-DHQ1 are shown as dashed lines. Relevant residues and water molecules and distances are shown and labeled. | ||
Unbiased, calculated electron density maps showed clear and high quality electron density for the enzyme-modified ligand molecule 4. The structure revealed that the addition adduct of the ε-amino group of the Lys170 residue and the ketone group in 4 was obtained and, more importantly, dehydration of the C3 hydroxyl group did not take place since clear electron density was observed for this hydroxyl group. The NZ atom of the modified Lys170 residue establishes a strong interaction with the conserved residue Asp114 via a water molecule (structural, W23) – an interaction that is found in all related crystal structures reported previously. The essential His143 residue interacts by hydrogen bonding with the C3 hydroxyl group of the modified ligand. In addition, the C3 hydroxyl and the C1 carboxylate groups of the modified ligand are linked via bridges involving water molecules (W81 and W104). The modified ligand is also anchored to the active site by the same type of electrostatic and hydrogen bonding interactions as the natural substrate, specifically, residues Gln236 and Ser232 (substrate-covering loop), and Arg213, Ser21, Arg48, Arg82 and Glu46.
Moreover, MD simulation studies carried out with the addition intermediate adduct St-DHQ1/4 clearly showed that the oxygen lone pairs of the C3 hydroxyl group would not be engaged by the guanidinium group of Arg82 (Fig. S3†). Thus, this group would rotate freely, and the guanidinium group of Arg82 would mainly interact with the C4 hydroxyl group of the modified ligand 4 instead of interacting with its C3 hydroxyl group. During the simulation, a large variability of the dihedral angle among the C2, C3, O3 and HO3 atoms of the modified ligand 4 is observed (Fig. S3C†). When the same analysis was carried out with the addition intermediate adduct obtained with the natural substrate, which has a hydroxyl group in C1 (Fig. S1C†), no rotation of the C3 hydroxyl group was observed as its oxygen lone pairs were held by Arg82 (Fig. 2D). Furthermore, the ε-nitrogen atom of the modified Lys170 by the modified natural substrate would be in an appropriate anti-periplanar arrangement for subsequent dehydration to afford the substrate-Schiff base. Thus, during the simulation the average value of the dihedral angle among the O3, C3, NZ and CE atoms of the modified substrate would be −61.8° (Fig. S4†). A less efficient anti-periplanar conformation was observed in the modified ligand 4, since the latter dihedral angle has a value of −68.1° in the crystal structure and an average value of −72.6° during the simulation (Fig. S4†). Taken together, the results of our structural and computational studies confirm our hypothesis about the key role of the C1 hydroxyl group in controlling the correct arrangement of the generated C3 hydroxyl group in the addition intermediate adduct for dehydration to afford the substrate-Schiff base.
 | ||
| Fig. 5 Crystal structures of St-DHQ1 covalently modified by 1–3. (A, B, and C) Unbiased electron density for the modified ligands 1 (A, violet), 2 (B, cyan), and 3 (C, orange), and their covalent attachment to Lys170 of St-DHQ1 (green). From the model obtained by molecular replacement and before the inclusion of the inhibitor molecule, refinement was performed to obtain unbiased density for the inhibitor molecule and other model changes. A maximum-likelihood weighted 2Fo − Fc map contoured at 1σ is shown up to 1.6 Å around the ligand molecule, the structural water molecule, and Lys170, His143 and Asp114 side chains (gray). The final model, including the ligand molecule, is superimposed onto the map. (D, E, and F) Interactions of the modified ligands 1 (violet), 2 (cyan), and 3 (orange) with St-DHQ1 (gray). Hydrogen bonding and electrostatic interactions between the ligands and St-DHQ1 are shown as dashed lines. Relevant residues are shown and labeled. | ||
Inhibition studies
The capacity of the reported hydroxylammonium derivatives to cause the irreversible inhibition of the DHQ1 enzyme from S. typhi (St-DHQ1) and S. aureus (Sa-DHQ1) was analyzed by incubation of both enzymes with ligands 1–3 for a 24 h period (PPB, pH 7.0, 25 °C). The activity was progressively determined by UV spectroscopy under the standard assay conditions using aliquots from the incubation samples and the control. The ligands 1–3 were found to be slow time-dependent irreversible inhibitors of the two enzymes (Fig. S5†). The inactivation proved to be more efficient for Sa-DHQ1 than for St-DHQ1. For the S. typhi enzyme, compound 2 proved to be the most efficient of the three ligands (4 h, ∼50% inactivation, ∼@1:120 enzyme/ligand ratio), whereas compound 1 was for Sa-DHQ1 (2.5 h, ∼50% inactivation, ∼@1:120 enzyme/ligand ratio).
Computational studies
The binding modes of ligands 1–3 in the active site of St-DHQ1 were first studied by molecular docking using the program GOLD 5.2.236 and further analyzed by MD simulation studies (100 ns). The monomer of the binary St-DHQ1/1–3 protein complexes in a truncated octahedron of water molecules obtained with the molecular mechanics force field AMBER was employed. These simulation studies were carried out considering the two possible protonation states of His143 (δ and dual). However, the latter possibility (dual) was ruled out because it caused an opening of the active site and a displacement of the ligands. The results revealed that the conserved Asp114 residue would be the main fragment responsible for the exquisite control of the arrangement of the leaving group and consequently of the methylene group at which covalent modification occurs (Fig. 6A–C). In particular, the hydroxyl group of the hydroxylammonium moiety would facilitate the establishment of a strong and stable hydrogen bonding interaction with the carboxylate group of Asp114. The average distance between Asp114 (OD1 atom) and the NH2O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) group (H04 atom) in 1–3 during the whole simulation was ∼1.6 Å in all cases (Fig. S6†). In addition, the hydrogen bonding interaction of the ligand side chain with the essential His143 would also greatly contribute towards freezing the conformation of the C3 side chain (Fig. S6†). Moreover, the average distances between His143 (NE2 atom) and the N2OH group (HZ atom) in 1–3 were calculated to be ∼2.5, 2.1 and 2.3 Å, respectively. For ligands 2–3, a strong and stable hydrogen bonding interaction with the structural water (average distance of ∼2.1 Å) is the third contact which fixes the conformation of the leaving group. This contact was not observed for ligand 1 which seems to be caused by intramolecular hydrogen bonding between the C3 hydroxyl group and the NH2OH group to provide a different arrangement of the NZ atom.
group (H04 atom) in 1–3 during the whole simulation was ∼1.6 Å in all cases (Fig. S6†). In addition, the hydrogen bonding interaction of the ligand side chain with the essential His143 would also greatly contribute towards freezing the conformation of the C3 side chain (Fig. S6†). Moreover, the average distances between His143 (NE2 atom) and the N2OH group (HZ atom) in 1–3 were calculated to be ∼2.5, 2.1 and 2.3 Å, respectively. For ligands 2–3, a strong and stable hydrogen bonding interaction with the structural water (average distance of ∼2.1 Å) is the third contact which fixes the conformation of the leaving group. This contact was not observed for ligand 1 which seems to be caused by intramolecular hydrogen bonding between the C3 hydroxyl group and the NH2OH group to provide a different arrangement of the NZ atom.
 | ||
| Fig. 6 Covalent modification mechanism of Lys170 by ligands 1–3. (A, B, and C) Proposed binding mode of ligands 1 (A, pink), 2 (B, cyan) and 3 (C, orange) in the active site of St-DHQ1 obtained by MD simulation studies. The pose of the ligands shown corresponds to that obtained after 90 ns of simulation. The side chains of the most relevant residues are shown and labeled. The residue Asp114 (green), which is key in the geometric control of the side chain of the ligand, the essential Lys170 (red), the essential His143 (yellow) and the catalytic water, as well as the most relevant polar interactions, are highlighted. (D, E, F, and G) QM/MM MD simulation studies for the covalent modification mechanism of St-DHQ1 by compound 2. Representative geometries of TS1 (D) and intermediate INT (E). Relevant residues and water molecules are shown and labeled. Key hydrogen bonding interactions and bonds broken/formed (distances included) are indicated as blue and red dashed lines, respectively. The residues Lys170 (orange) and His143 (yellow) are highlighted. Geometries were taken from the free energy surface (F). (G) Free energy profile obtained using umbrella sampling simulations at the SCC-DFTB/ff14SB level for the whole reaction. | ||
In an effort to get an insight into how the reported ligands cause the covalent modification of the catalytic lysine residue, a combined quantum mechanics/molecular mechanics (QM/MM) umbrella sampling simulation study was carried out. Modelling of reactions in enzymes with QM/MM methods can provide knowledge in atomic detail about the mechanism, the key interactions of the ligands, reaction intermediate(s) and transition state(s).37,38 By using these methods, the electronic rearrangements involved in the breaking/making of chemical bonds (QM) are studied in a dynamic environment (MM) which mimics the conformational changes of the enzyme occurring during the reaction, which are also important for the reaction energetics. In addition, by using umbrella sampling techniques, the exploration of regions along the reaction coordinate which would otherwise have insufficient sampling is achieved. These QM/MM umbrella sampling simulation studies were performed with compound 2 and diverse geometries on the non-covalent binary St-DHQ1/2 complex were obtained by MD simulation studies. Three possible reaction coordinates were explored: (i) breakdown of the C–N bond in 2; (ii) formation of the C(2)–N(K170) bond; and (iii) a linear combination of (i) minus (ii). Two possible protonation states of His143 (δ and dual) were employed. However, the latter possibility (dual) was ruled out because it caused an opening of the active site. The best results were obtained with the two-dimensional US (iii) (see the ESI† for full details). The resulting free energy profile at the SCC–DFTB/ff14SB level indicates that the SN2 nucleophilic substitution by Lys170 with the release of NH2OH would be feasible, with an energy barrier of 27.2 kcal mol−1 for the TS (TS1), which would be the rate-determining step (Fig. 6D–G). The reaction would involve the formation of an intermediate INT with an energy barrier of 11.8 kcal mol−1 which would undergo a subsequent deprotonation by neutral His143 having a very low reaction barrier (0.9 kcal mol−1 relative to INT, TS2) (Fig. S7†).
Conclusions
In summary, we have demonstrated that quinic acid-based hydroxylammonium derivatives can be used for the selective covalent modification of the catalytic lysine residue of the DHQ1 enzyme, which is a promising target for anti-virulence drug discovery. This was evidenced by the resolution of the crystal structures of DHQ1 from S. typhi (resolution range: 1.08–1.25 Å) chemically modified by the reported ligands 1–3 showing that the modified ligands are linked to the lysine residue through an amine group. The results obtained with compounds 2 and 3 reveal that the presence of a hydroxyl group in position C3 of the reported scaffold is not required to achieve covalent modification allowing the use of quite simple ligands such as compound 3. The results from the MD and two-dimensional QM/MM umbrella sampling simulation studies suggested that the covalent modification mechanism might take place by the direct nucleophilic attack of the ε-amino group of the lysine residue with the release of NH2OH followed by deprotonation of the lysine adduct by the essential histidine. To our knowledge, this is the first example of hydroxylammonium derivatives that cause the specific covalent modification of a catalytic and sterically inaccessible lysine residue of an enzyme. These studies might open up new opportunities for the development of novel lysine-targeted irreversible inhibitors bearing a hydroxylammonium moiety as a latent electrophile.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competiveness (SAF2016-75638-R), the Xunta de Galicia [Centro singular de investigación de Galicia accreditation 2016-2019 (ED431G/09) and ED431B 2018/04] and the European Union (European Regional Development Fund – ERDF) is gratefully acknowledged. MM and EL thank the Spanish Ministry of Education, Culture and Sport and the Xunta de Galicia for their respective FPU and postdoctoral fellowships. We are grateful to the ALBA synchrotron (Barcelona, Spain) for the provision of beam time and to the Centro de Supercomputación de Galicia (CESGA) for use of the Finis Terrae computer.Notes and references
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- D. S. Johnson, E. Weerapana and B. F. Cravatt, Future Med. Chem., 2010, 2, 949–964 CrossRef CAS PubMed.
- D. C. Swinney, Nat. Rev. Drug Discovery, 2004, 3, 801–808 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–377 CrossRef CAS PubMed.
- K. Sanderson, Nat. Rev. Drug Discovery, 2013, 12, 649–651 CrossRef CAS PubMed.
- J. A. Blair, D. Rauh, C. Kung, C.-H. Yun, Q.-W. Fan, H. Rode, C. Zhang, M. J. Eck, W. A. Weiss and K. M. Shokat, Nat. Chem. Biol., 2007, 3, 229–238 CrossRef CAS PubMed.
- J. Singh, R. C. Petter and A. F. Kluge, Curr. Opin. Chem. Biol., 2010, 14, 475–480 CrossRef CAS PubMed.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- C. Jöst, C. Nitsche, T. Scholz, L. Roux and C. D. Klein, J. Med. Chem., 2014, 57, 7590–7599 CrossRef PubMed.
- M. S. Cohen, C. Zhang, K. M. Shokat and J. Taunton, Science, 2005, 308, 1318–1321 CrossRef CAS PubMed.
- R. Ekkebus, S. I. van Kasateren, Y. Kulathu, A. Scholten, I. Berlin, P. P. Geurink, A. de Jong, S. Goerdayal, J. Neefjes, A. J. R. Heck, D. Komander and H. Ovaa, J. Am. Chem. Soc., 2013, 135, 2867–2870 CrossRef CAS PubMed.
- M. Hagel, D. Niu, T. St Martin, M. P. Sheets, L. Qiao, H. Bernard, R. M. Karp, Z. Zhu, M. T. Labenski, P. Chaturvedi, M. Nacht, W. F. Westlin, R. C. Petter and J. Singh, Nat. Chem. Biol., 2011, 7, 22–24 CrossRef CAS PubMed.
- B. R. Lanning, L. R. Whitby, M. M. Dix, J. Douhan, A. M. Gilbert, E. C. Hett, T. O. Johnson, C. Joslyn, J. C. Kath, S. Niessen, L. R. Roberts, M. E. Schnute, C. Wang, J. J. Hulce, B. Wei, L. O. Whitely, M. M. Hayward and B. F. Cravatt, Nat. Chem. Biol., 2014, 10, 760–767 CrossRef CAS PubMed.
- D. W. Fry, A. J. Bridges, W. A. Denny, A. Doherty, K. D. Greis, J. L. Hicks, K. E. Hook, P. R. Keller, W. R. Leopold, J. A. Loo, D. J. McNamara, J. M. Nelson, V. Sherwood, J. B. Smaill, S. Trumpp-Kallmeyer and E. M. Bobrusin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12022–12027 CrossRef CAS PubMed.
- A. H. Chan, W.-G. Lee, K. A. Spasov, J. A. Cisneros, S. N. Kudalkar, Z. O. Petrova, A. B. Buckingham, K. S. Anderson and W. L. Jorgensen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9725–9730 CrossRef CAS PubMed.
- W. Zhou, D. Ercan, L. Chen, C.-H. Yun, D. Li, M. Capelletti, A. B. Cortot, L. Chirieac, R. E. Iacob, R. Padera, J. R. Engen, K.-K. Wong, M. J. Eck, N. S. Gray and P. A. Jänne, Nature, 2009, 462, 1070–1074 CrossRef CAS PubMed.
- J. M. Osterm, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef PubMed.
- J. Pettinger, K. Jones and M. D. Cheeseman, Angew. Chem., Int. Ed., 2017, 56, 15200–15209 CrossRef CAS PubMed.
- Q. Zhao, X. H. Ouyang, X. B. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- N. P. Grimster, S. Connelly, A. Baranczak, J. Dong, L. B. Krasnova, K. B. Sharpless, E. T. Powers, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 5656–5668 CrossRef CAS PubMed.
- E. Anscombe, E. Meschini, R. Mora-Vidal, M. P. Martin, D. Staunton, M. Geitmann, U. H. Danielson, W. A. Stanley, L. Z. Wang, T. Reuillon, B. T. Golding, C. Cano, D. R. Newell, M. E. M. Noble, S. R. Wedge, J. A. Endicott and R. J. Griffin, Chem. Biol., 2015, 22, 1159–1164 CrossRef CAS PubMed.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS PubMed.
- P. A. Smith, et al. , Nature, 2018, 561, 189–194 CrossRef CAS PubMed.
- P. Martín-Gago and C. A. Olsen, Angew. Chem., Int. Ed., 2019, 58, 957–966 CrossRef PubMed.
- L. H. Jones, ACS Med. Chem. Lett., 2018, 9, 584–586 CrossRef CAS PubMed.
- C. González-Bello, Future Med. Chem., 2015, 7, 2371–2383 CrossRef PubMed.
- D. G. Gourley, A. K. Shrive, I. Polikarpov, T. Krell, J. R. Coggins, A. R. Hawkins, N. W. Isaacs and L. Sawyer, Nat. Struct. Biol., 1999, 6, 521–525 CrossRef CAS PubMed.
- A. Karnell, P. D. Cam, N. Verma and A. A. Lindberg, Vaccine, 1993, 11, 830–836 CrossRef CAS PubMed.
- M. Malcova, D. Karasova and I. Rychlik, FEMS Microbiol. Lett., 2009, 291, 44–49 CrossRef CAS PubMed.
- D. A. Dilts, I. Riesenfeld-Orn, J. P. Fulginiti, E. Ekwall, C. Granert, J. Nonenmacher, R. N. Brey, S. J. Cryz, K. Karlsson, K. Bergman, T. Thompson, B. Hua, A. H. Brückner and A. A. Lindberg, Vaccine, 2000, 18, 1473–1484 CrossRef CAS PubMed.
- L. Tizón, M. Maneiro, A. Peón, J. M. Otero, E. Lence, S. Poza, M. J. van Raaij, P. Thompson, A. R. Hawkins and C. González-Bello, Org. Biomol. Chem., 2015, 13, 706–716 RSC.
- C. González-Bello, L. Tizón, E. Lence, J. M. Otero, M. J. van Raaij, M. Martinez-Guitian, A. Beceiro, P. Thompson and A. R. Hawkins, J. Am. Chem. Soc., 2015, 137, 9333–9343 CrossRef PubMed.
- B. Blanco, V. Prado, E. Lence, J. M. Otero, C. Garcia-Doval, M. J. van Raaij, A. Llamas-Saiz, H. Lamb, A. R. Hawkins and C. González-Bello, J. Am. Chem. Soc., 2013, 135, 12366–12376 CrossRef CAS PubMed.
- C. Kleanthous, M. Reilly, A. Cooper, S. Kelly, N. C. Price and J. R. Coggins, J. Biol. Chem., 1981, 266, 10893–10898 Search PubMed.
- https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ (last access March 15, 2019).
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- M. W. Van der Kamp and A. J. Mulholland, Biochemistry, 2003, 42, 2708–2728 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7 and one table, full details of the Experimental section (synthesis, protein crystallization and structural determination, enzyme assays and computational studies) and NMR spectra. Coordinates and structure factors are available from the Protein Data Bank with accession codes 6H5C, 6H5D, 6H5G, and 6H5J. See DOI: 10.1039/c9qo00453j |
| This journal is © the Partner Organisations 2019 |