
A controlled blue-shift in meso-nitrogen aryl fused DIPY and BODIPY skeletons†

Abstract
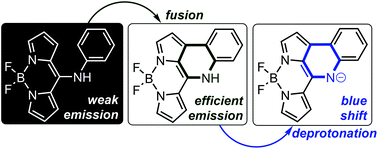
meso-Nitrogen substituted dipyrromethenes (DIPYs) and their boron(III) complexes (BODIPYs) were obtained by the method of nucleophilic aromatic substitution. The following intramolecular fusion with the formation of a 3H-[2,3-c]quinoline skeleton extends delocalisation, but shifts the absorption and emission hypsochromically. A further blue shift has been observed for a deprotonated form of the fused system. A nitrogen atom introduced into the system can be treated as a switching factor that controls the delocalisation significantly influencing the emission by shifting between the available amine and imine tautomeric forms.
 Please wait while we load your content...
Please wait while we load your content...

