
Gold-catalyzed heterocyclic syntheses through α-imino gold carbene complexes as intermediates†
 *a
and
Javier
Santamaría
*a
*a
and
Javier
Santamaría
*a
Abstract
Gold carbene complexes have been recognized as common intermediates in gold-catalyzed organic syntheses. In this field, α-imino gold carbene complexes, in the last few years, have emerged as valuable intermediates toward the synthesis of N-heterocycles. This review is dedicated toward formulating a comprehensive compilation of the different methodologies for heterocyclic synthesis, postulating the participation of α-imino gold carbene complexes as intermediates. In addition to the scarce examples involving the direct formation of α-imino diazo compounds from gold decomposition, the use of nitrogenated nucleophiles, through an initial attack on gold-activated alkynes followed by gold retrodonation and expulsion of a leaving group, constitutes the most commonly employed strategy for achieving this target. This review has been divided into different sections as follows according to the type of N-nucleophile used: azides, aza-ylides, 2H-azirines, isoxazoles and their derivatives, indazoles, and triazapentalenes. A large number of heterocycles, ranging from five- to seven-membered rings, have been efficiently synthesized following this methodology.
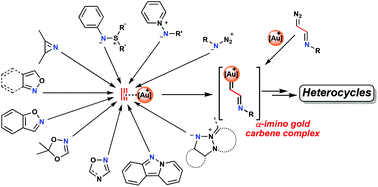
- This article is part of the themed collections: 2019 Organic Chemistry Frontiers Review-type Articles and In celebration of Julius Rebek’s 75th Birthday
 Please wait while we load your content...
Please wait while we load your content...

