
Photocatalytic borylcyclopropanation of α-boryl styrenes†

Abstract
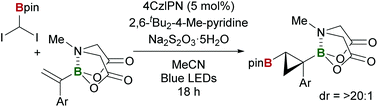
A diastereoselective borylcyclopropanation of α-MIDA-boryl styrenes with 1,1-diiodoborylmethane is developed using 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) as a catalyst under visible-light irradiation. The scope of this photocatalytic method is explored, and the utility of the resulting doubly borylated cyclopropanes is demonstrated by the selective transformation of one of the two boryl groups.
- This article is part of the themed collection: In celebration of Julius Rebek’s 75th Birthday
 Please wait while we load your content...
Please wait while we load your content...

