
A computational study on ruthenium-catalyzed [4 + 1] annulation via C–H activation: the origin of selectivity and the role of the internal oxidizing group†
Bing
Lian
a,
Lei
Zhang
 ab and
De-Cai
Fang
*a
ab and
De-Cai
Fang
*a
aCollege of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: dcfang@bnu.edu.cn
bTianjin Engineering Technology Center of Chemical Wastewater Source Reduction and Recycling, School of Science, Tianjin Chengjian University, Tianjin 300384, P. R. China
First published on 7th March 2019
Abstract
We report a density functional theory study on ruthenium-catalyzed [4 + 1] annulation of benzamides with propargyl alcohols via C–H activation. The plausible catalytic cycles involve N–H deprotonation, C–H activation, alkyne insertion, β-H elimination, enol–keto tautomerism, oxidative addition, C–N reductive elimination, protonation, and catalyst recovery. Importantly, the preference of the [4 + 1] annulation over the [4 + 2] annulation is discussed in detail. Alkynes bearing a β-H group and a hydroxyl group are necessary for β-H elimination and enol–keto tautomerism, which lead to the formation of a [4 + 1] product. In contrast, alkynes without a β-H group or a hydroxyl group could only lead to [4 + 2] annulation. Additionally, this article addresses the key role of the internal oxidizing group, which is responsible for converting RuII into RuIV and thus favoring the following C–N reductive elimination. Hence, the studied mechanism of [4 + 1] annulation is a novel RuII/IV/II pathway rather than a RuII/0/II pathway.
1. Introduction
Transition-metal-catalyzed cycloaddition reactions via C–H activation are useful synthetic tools in modern organic chemistry, because of their atom-economy and versatile advantages.1 Aromatic compounds bearing a suitable directing group are important raw materials in transition-metal-catalyzed C–H functionalizations, because the formation of cyclometalated intermediates enables ortho-selective cross-couplings.2 In general, C–H activation only serves as the initial step of whole catalytic cycles, and a coupling partner would subsequently react with the cyclometalated intermediate to furnish the desired product. In some cases, separation of the product and regeneration of the catalyst need the help of a stoichiometric amount of an external oxidant.3 To avoid the waste of the oxidant and the use of harsh oxidative conditions, a series of internal oxidizing groups also playing the role of directing groups have been developed and utilized in recent years.4Transition-metal-catalyzed intermolecular annulations of aromatic compounds with internal alkynes, alkenes and allenes5 are quite different from concerted pericyclic reactions, such as the Diels–Alder reactions.6 Internal alkynes have been widely used as two-carbon synthons in these annulations, such as [2 + 2],7 [3 + 2],8 [4 + 2],9 and [5 + 2]10 annulations, but those serving as one-carbon synthons have seldom been reported.11
In 2017, Liu and co-workers reported a novel synthetic method for ruthenium-catalyzed redox-neutral [4 + 1] annulation of benzamide (1a) with propargyl alcohol (2a) (see Scheme 1), with the latter species serving as a one-carbon synthon rather than a two-carbon synthon found in the [4 + 2] annulations of arenes with alkynes.12 Under the optimal reaction conditions shown in Scheme 1, the formation of the formal [4 + 1] annulation product (3a) was preferred over that of the [4 + 2] annulation product (4a) with a ratio of 98/2, indicating a high chemoselectivity in favor of the [4 + 1] annulation. It was proposed that the cleavage of the N–O bond in 1a should be responsible for reoxidation of the catalyst, which avoided the undesired use of an external oxidant.
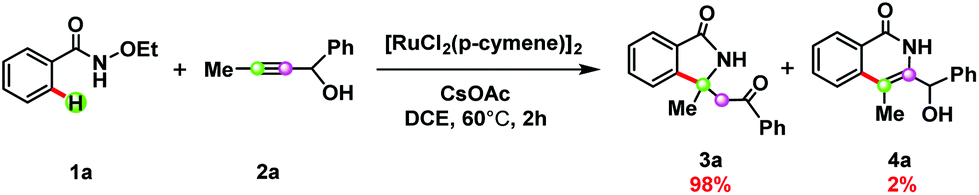 | ||
| Scheme 1 Ruthenium-catalyzed redox-neutral [4 + 1] annulation of benzamides with propargyl alcohols. | ||
Owing to the increasing importance of ruthenium-catalyzed annulation reactions and the lack of mechanistic details on the intermolecular [4 + 1] annulation of arenes with alkynes, density functional theory (DFT) studies are employed to explore the plausible mechanisms leading to the products 3a and 4a. To explain why the production of 3a is biased, the possible mechanisms of the [4 + 1] annulation and [4 + 2] annulation are compared. In addition, the role of the internal oxidizing group will be unveiled in the end.
2. Computational details
Calculations were carried out using the Gaussian 09 program package.13 Geometries of reactants, intermediates, transition states, and products were first optimized by the Becke three-parameter Lee–Yang–Parr (B3LYP) functional method,14 with the TZP-DKH basis set15 used for all elements, in which d functions of H atoms, f functions of C, O, and N atoms, and g functions of Ru atoms were removed (denoted as TZP-DKH(-dfg)). The solvent effect of dichloroethane (ε = 10.125) was considered by the polarized continuum model (PCM)16 using our defined IDSCRF17 radii. The effects of the dispersion interaction were calculated by employing the B3LYP-D3(BJ)18 functional method in both geometrical optimization and energetic determination. The solution translational entropy corrections have been carried out using our THERMO program.19 Three-dimensional structures were illustrated using CYLview.203. Results and discussion
3.1. Proposed catalytic cycles
Previous mechanistic studies on ruthenium-catalyzed C–H activation proposed that the dimeric catalyst [RuCl2(p-cymene)]2 can dissociate into two monomers Ru(OAc)2(p-cymene) with the help of CsOAc.21 Therefore, Ru(OAc)2(p-cymene) should serve as the active catalyst.A general mechanistic picture for ruthenium-catalyzed annulation of 1a with 2a is given in Scheme 2. Initially, N–H deprotonation of 1a by Ru(OAc)2(p-cymene) gives the intermediate A, which is followed by C–H activation on the ortho position to generate the cyclometalated intermediate B. Then 2a replaces the neutral acetic acid ligand and inserts into the formed Ru–aryl bond in B to produce the seven-membered ring intermediate C. Afterwards, there are two competing reaction pathways leading to different annulation products. On the one hand, the hydrogen shift and the ruthenium shift give the intermediate D, and it then undergoes an oxidative addition via the elimination of EtOH to deliver the RuIV species E; C–N reductive elimination generates the key [4 + 1] annulation intermediate F, which leads to the desired [4 + 1] product 3a (blue pathway in Scheme 2). On the other hand, C–N reductive elimination gives the six-membered ring complex M, which can undergo an oxidative addition via the N–O cleavage to form N, and protonation by acetic acid yields the [4 + 2] annulation product 4a (red pathway in Scheme 2). The detailed reaction mechanisms for all of the typical reaction steps will be discussed in the following subsections.
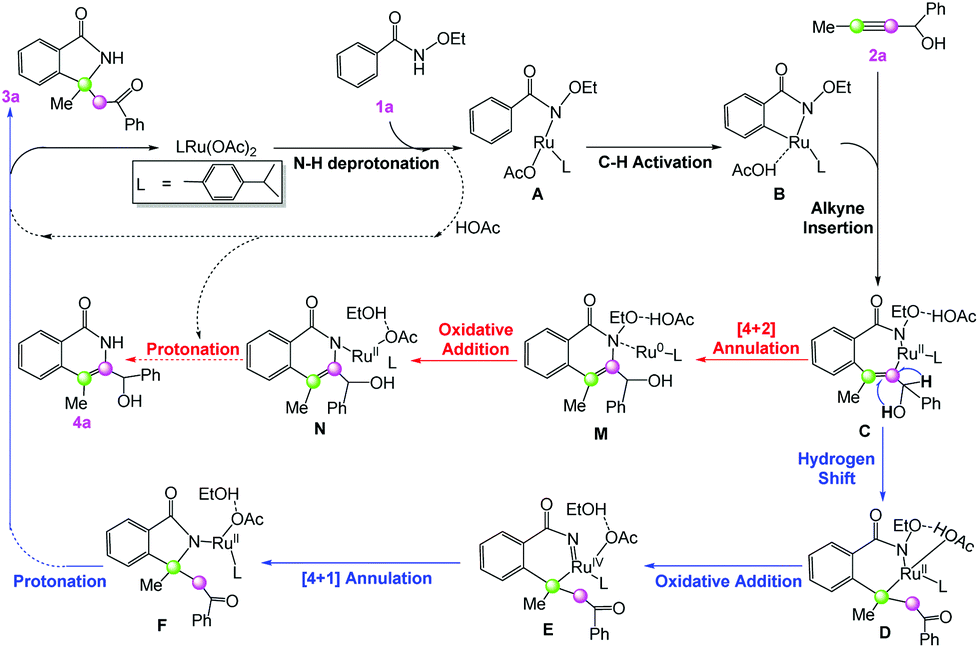 | ||
| Scheme 2 Possible catalytic cycles for ruthenium-catalyzed annulation of benzamides with propargyl alcohols: [4 + 1] annulation (in blue) and [4 + 2] annulation (in red). | ||
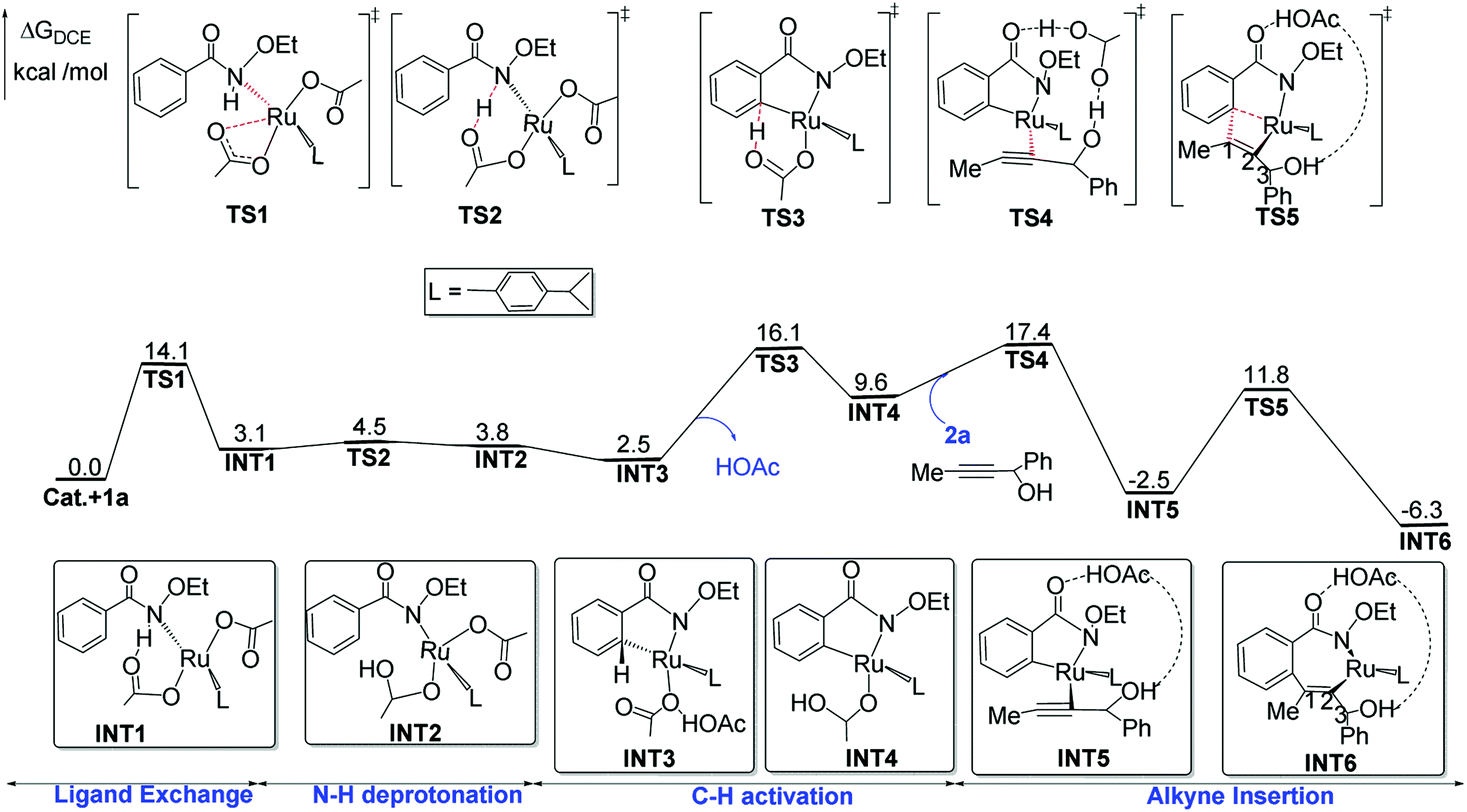 | ||
| Fig. 1 Proposed C–H activation and alkyne insertion mechanisms of ruthenium-catalyzed annulation of benzamides with propargyl alcohols, along with ΔG (in kcal mol−1) determined at the B3LYP-D3+IDSCRF/TZP-DKH(-dfg) level of theory in DCE. | ||
Then, a ligand exchange between 2a and acetic acid viaTS4 gives the intermediate INT5, and alkyne insertion viaTS5 generates the seven-membered ruthenacyclic intermediate INT6. In the structure of INT6 (see Fig. 2), a molecule of acetic acid serves as the bridge linking the hydroxyl group of 2a and the carbonyl group of 1a through hydrogen bonds, leading to the observation that the β-H atom, rather than the hydroxyl oxygen, coordinates to the ruthenium center. The p-cymene ligand should coordinate to the ruthenium center throughout the entire reaction, as proposed by some previous mechanistic studies on Ru(OAc)2(p-cymene) catalyzed C–H activation.23
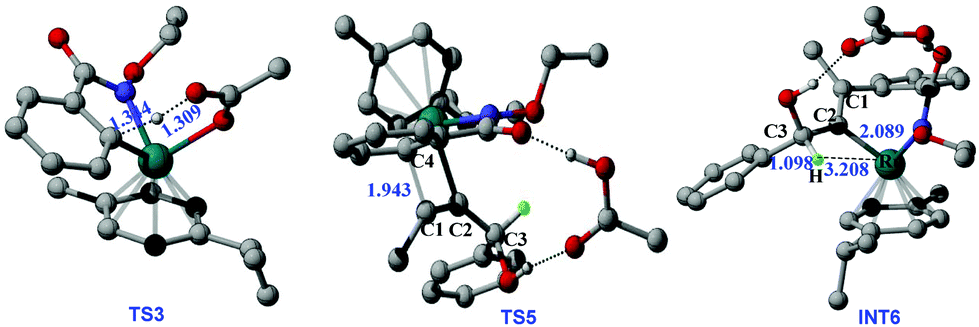 | ||
| Fig. 2 Geometric structures for selected transition states and intermediates of C–H activation and alkyne insertion processes. In 3D structures, hydrogen atoms not for proton shift are omitted for clarity (distance in Å). | ||
Fig. 1 provides the relative free-energies calculated at the B3LYP-D3+IDSCRF/TZP-DKH(-dfg) level of theory in DCE. The complexation between 1a and Ru(OAc)2(p-cymene) has a free-energy barrier of 14.1 kcal mol−1. The following N–H deprotonation step is free-energy endergonic by 3.1 kcal mol−1. The intramolecular ligand exchange step, INT2 → INT3, is exergonic by 1.3 kcal mol−1. The C–H activation transition state TS3 is 16.1 kcal mol−1 above the initial point and then the reaction releases free-energies by 6.5 kcal mol−1 to form INT4. The following intermolecular ligand substitution between 2a and acetic acid viaTS4 has a free-energy barrier of 7.8 kcal mol−1 and releases free energies by 19.9 kcal mol−1 to form INT5. The alkyne insertion transition state TS5 is computed to be 11.8 kcal mol−1 above the initial point and the generated seven-membered ruthenacyclic intermediate INT-6 lies at 6.3 kcal mol−1 in free-energy below the starting reactants.
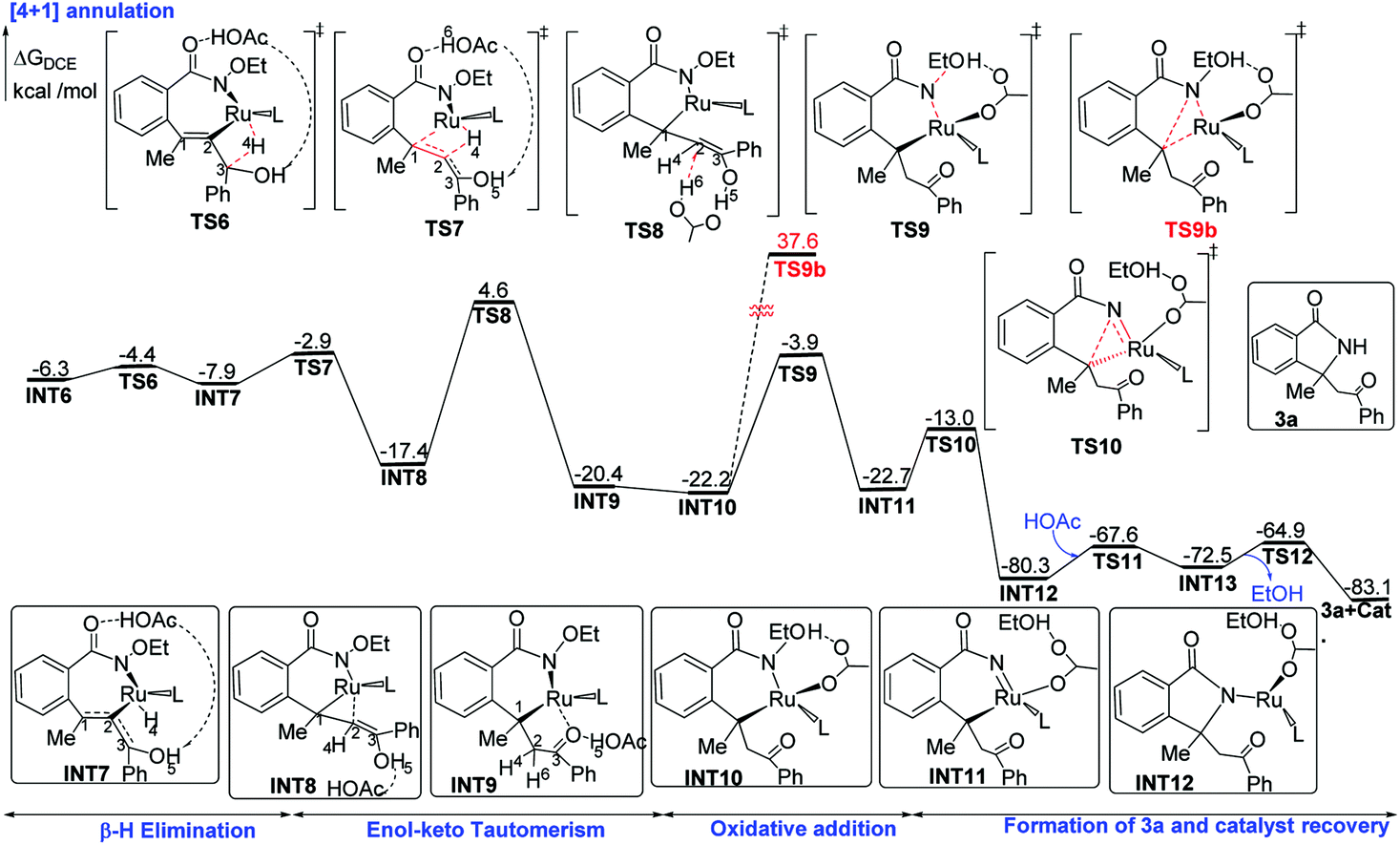 | ||
| Fig. 3 Proposed mechanisms of ruthenium-catalyzed [4 + 1] annulation of benzamides with propargyl alcohols, along with ΔG (in kcal mol−1) determined at the B3LYP-D3+IDSCRF/TZP-DKH(-dfg) level of theory in DCE. | ||
The migratory insertion transition state TS7 indicates that the H4 atom shifts from ruthenium to C2, and simultaneously, ruthenium shifts from C2 to C1. INT8 has a weak π-coordination of Ru–C2. The transformation from INT6 to INT8 is strongly exergonic by 11.1 kcal mol−1. According to the hydrogen shift transition state TS8, the H6 atom of acetic acid shifts to C2, and meanwhile the H5 atom of the hydroxyl group shifts to acetic acid, that is acetic acid serves as the bridge for hydrogen shift. Then, the C2 atom changes from sp2 to sp3 hybridization, resulting in the cleavage of the Ru–C2 bond and the formation of the Ru–C1 bond. These transformation steps change the seven-membered ruthenacyclic complex to the six-membered complex INT9. It is proposed that the successive β-H elimination and hydroxyl-induced enol–keto tautomerism lead to the unprecedented [4 + 1] annulation.
The acetic acid forms an O–H⋯O hydrogen bond with the internal oxidizing directing group and coordinates to the ruthenium center to generate the intermediate INT10. This conformational transformation step is exergonic in free-energy by 1.8 kcal mol−1. The internal oxidizing group N–OEt abstracts a proton from acetic acid viaTS9 with the cleavage of N–OEt to form the RuIV species INT11. Our calculation results show that this oxidative addition has a free-energy barrier of 18.3 kcal mol−1 and is exothermal by 0.5 kcal mol−1. Then, the C–N reductive elimination via the three-center transition state TS10 affords the [4 + 1] annulation complex INT12, which recovers the oxidation state of ruthenium from +4 to +2. The reductive elimination step from INT11 to INT12 needs a low free-energy barrier of 9.7 kcal mol−1 and is extremely exergonic by 57.6 kcal mol−1. Afterwards, an intramolecular ligand substitution gives INT-13, which finally undergoes protonation via the transition state TS-12 to liberate the desired [4 + 1] product 3a and regenerate the catalyst Ru(OAc)2(p-cymene).
According to the studied reaction pathway, the participation of the internal oxidizing group and the consequent occurrence of RuIV precede the C–N reductive elimination. However, experimental researchers proposed that the C–N reductive elimination possibly occurred from RuII species without the role of the oxidizing group.12 We have considered this mechanistic proposal and designed a C–N reductive elimination step from INT10 to TS9b, which is required to overcome a much higher barrier of 59.8 kcal mol−1 and thus should be quite challenging (see below for detailed contents). Therefore, the internal oxidizing group plays a key role in promoting the desired C–N bond coupling, and the formation of a RuIV precursor should be indispensable.
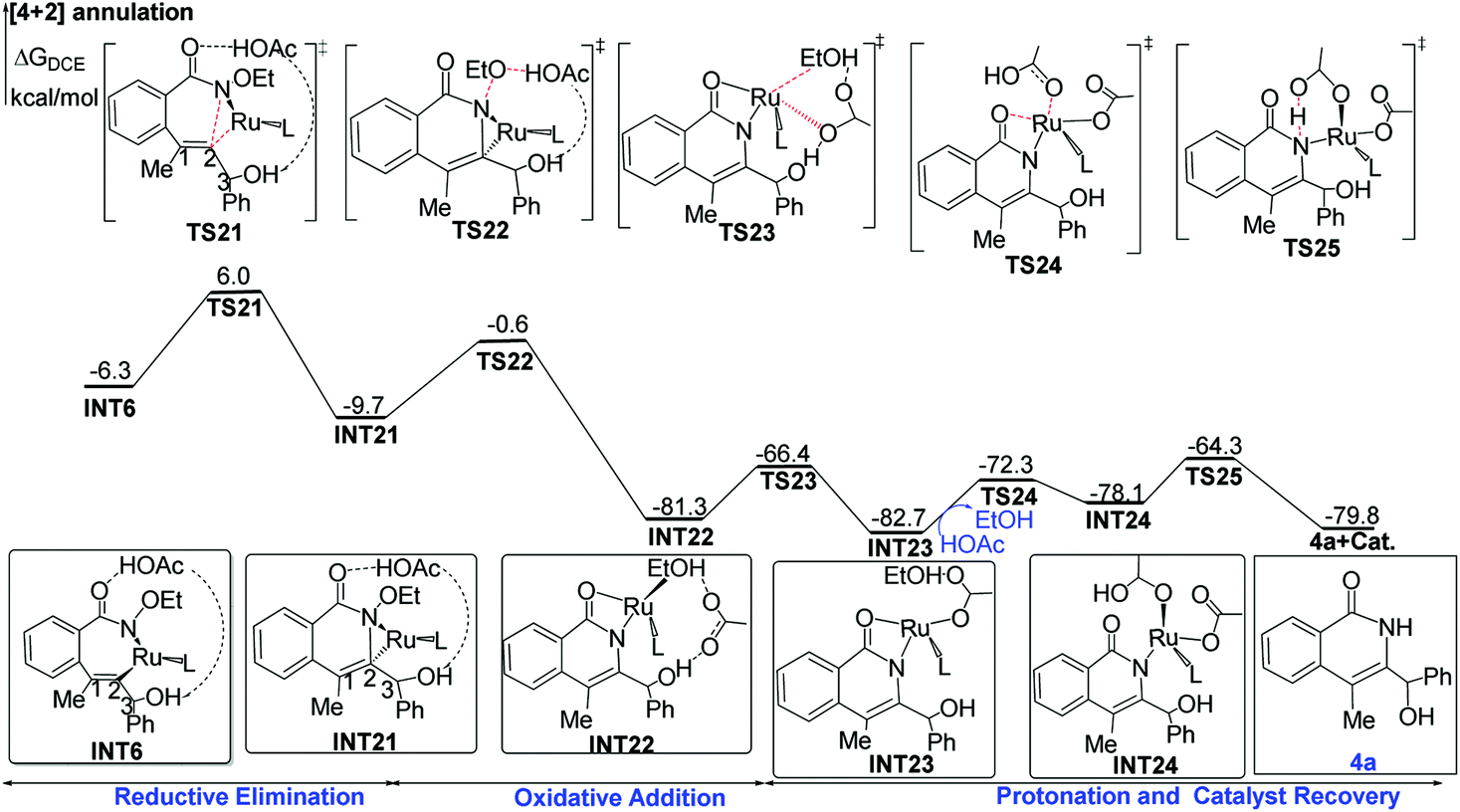 | ||
| Fig. 4 Proposed mechanisms of ruthenium-catalyzed [4 + 2] annulation of benzamides with propargyl alcohols, along with ΔG (in kcal mol−1) determined at the B3LYP-D3+IDSCRF/TZP-DKH(-dfg) level of theory in DCE. | ||
The cleavage of the N–O bond with the assistance of acetic acid viaTS22 forms the RuII-species INT22, and it is extremely exothermal by 80.7 kcal mol−1 from INT21 to INT22. Subsequently, acetate replaces EtOH to coordinate to the ruthenium center, leading to the more stable intermediate INT23 with release of free-energies by 1.4 kcal mol−1. Finally, intramolecular substitution and protonation yield the [4 + 2] product 4a and regenerate the catalyst.
3.2. Competition of [4 + 1] and [4 + 2] annulations
INT6 is the branch point for [4 + 2] and [4 + 1] annulations. The entire free-energy profiles of the [4 + 2] annulation and [4 + 1] annulation are comparatively depicted in Fig. S1,† the part that reflects chemoselectivity is given in Fig. 5.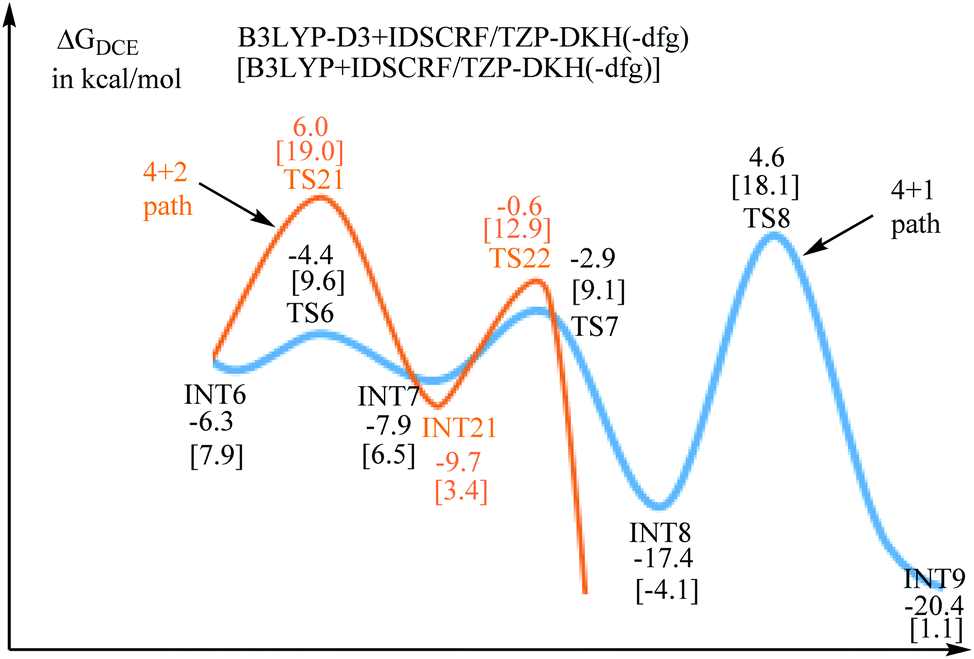 | ||
| Fig. 5 Comparison of the [4 + 2] annulation (in orange) and [4 + 1] annulation (in blue). | ||
The Curtin–Hammett principle stated that the product distribution reflects the difference in free energy between the two rate-limiting transition states (RLTS).25 From the steady-state approximation,26 the RLTS of the [4 + 1] path is TS8 with a free-energy barrier of 10.9 and 10.2 kcal mol−1 relative to INT6 at B3LYP-D3+IDSCRF/TZP-DKH(-dfg) and B3LYP+IDSCRF/TZP-DKH(-dfg) levels, respectively. However, the RLTS of the [4 + 2] path is TS21 with the corresponding free-energy barriers of 12.3 and 11.1 kcal mol−1, respectively.
Therefore, TS8 is ca. 1.4 (with D3 correction) or 0.9 kcal mol−1 (without D3 correction) more stable than TS21 in terms of free energy barriers, indicating that the [4 + 1] product should be more favorable than the [4 + 2] product, in good agreement with the control experiments (3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a = 98:2) by Liu and coworkers.12
:4a = 98:2) by Liu and coworkers.12
There are two factors stabilizing TS8. Firstly, the presence of the hydroxyl group gives the possibility of enol–keto tautomerism. Secondly, owing to the delocalized π system of acetate, the resonance promotes the H5 atom split from the hydroxyl group and facilitated H6 transfer to the C2 atom.
The processes from INT6 to INT9 are β-H elimination and enol–keto tautomerism, while the use of 2b or 2c instead of 2a failed to furnish the corresponding [4 + 1] annulation product 3b or 3c (see Scheme 3),5e,12 indicating that internal alkynes with a β-H group and a hydroxyl group27 should be essential for the formation of the [4 + 1] product. Species 2b does not possess a β-H and hence could only lead to [4 + 2] annulation. The lack of a hydroxyl group in 2c would prevent the [4 + 1] annulation pathway from INT8 and make it return to the [4 + 2] annulation pathway. The free energy barrier of the [4 + 2] annulation for the production of 4b and 4c is provided in the ESI.† In other words, both β-H elimination and enol–keto tautomerism induced by the hydroxyl group are the key factors for the formation of the precursor of the [4 + 1] product.
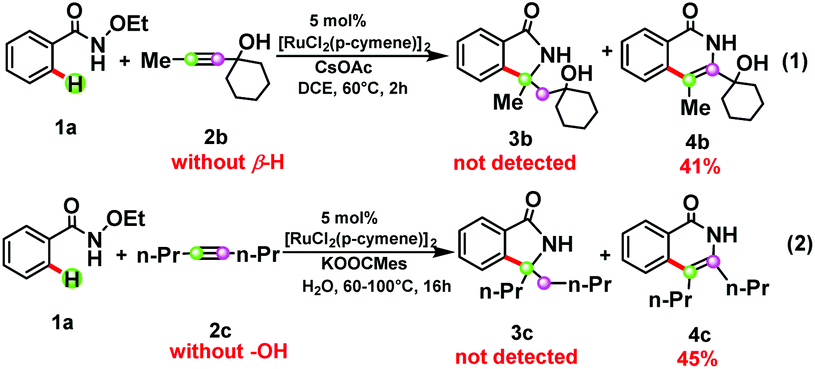 | ||
| Scheme 3 The dependence of alkynes on the selectivity between the [4 + 1] annulation and [4 + 2] annulation. | ||
3.3. Comparison of RuII/IV/II and RuII/0/II catalytic cycles
Some ruthenium-catalyzed transformations involve the change of the oxidation state due to the role of the internal oxidizing group. We found that the plausible pathway of the [4 + 1] annulation is a RuII/IV/II catalytic cycle, while that of the [4 + 2] annulation is a RuII/0/II catalytic cycle. Fig. 6 summarizes the key results for the possible RuII/IV/II and RuII/0/II catalytic cycles of the [4 + 1] and [4 + 2] annulation, namely (1) C–N reductive elimination from INT6 forming the [4 + 2] product; (2) rupture of the N–OEt bond with the help of an outer shell acetic acid forming the [4 + 2] product; (3) reductive elimination from INT10 forming the [4 + 1] product; (4) rupture of the N–OEt bond with the help of an inner shell acetic acid forming the [4 + 1] product.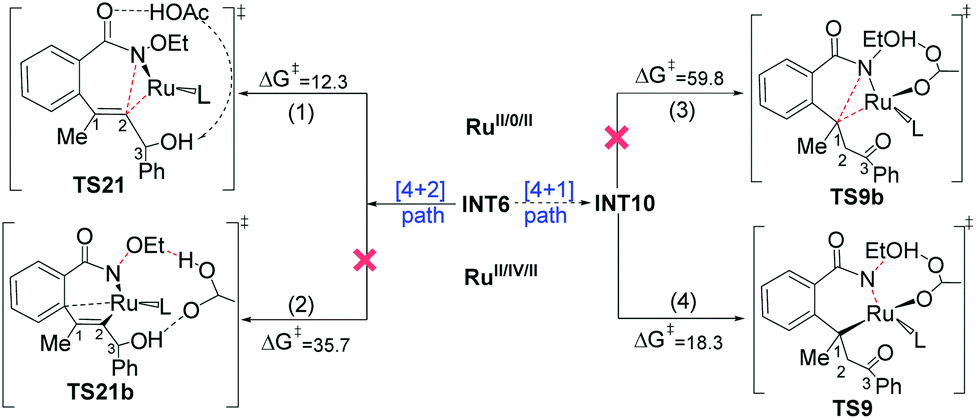 | ||
| Fig. 6 The sequence of reductive elimination and oxidative addition and the role of the internal oxidizing group in the [4 + 2] annulation (left) and [4 + 1] annulation (right), respectively. Free-energy data (in kcal mol−1) are determined at the B3LYP-D3+IDSCRF/TZP-DKH(-dfg) level of theory in DCE. | ||
The first route is the direct C–N reductive elimination from INT6viaTS21, leading to the change of the oxidative state of Ru from +2 to 0, and it has a low free-energy barrier of 12.3 kcal mol−1. In the second route, the formed acetate group and EtOH cannot coordinate to RuIV due to steric hindrance, and such a charge separation process has a high free-energy barrier of 35.7 kcal mol−1. In the third pathway, the C1–N reductive elimination at RuII to form Ru0 and a [4 + 1] product has quite a high free-energy barrier of 59.8 kcal mol−1, since C1 in INT10 is saturated (sp3-hybrid) and the C1–N bond formation would result in the over-saturation of C1 in TS9b. In the last route, the ruthenium center in TS9 changes from RuII to RuIV, but the formed Ru–O bond could compensate the energy cost required by the rupture of the N–O bond, which has been evoked in some previous computational studies on Rh and Pd systems.28
In summary, the [4 + 1] annulation follows a RuII/IV/II pathway, while the [4 + 2] annulation preferably follows a RuII/0/II pathway.
4. Conclusions
DFT calculations have been carried out to elucidate the mechanisms and selectivity of a ruthenium-catalyzed redox-neutral [4 + 1] annulation of benzamides with internal alkynes via C–H activation. The unprecedented [4 + 1] annulation is confirmed to be the plausible pathway and to proceed through a multi-step process: N–H deprotonation and C–H activation via CMD transition states, alkyne insertion, β-H elimination, enol–keto tautomerism, oxidative addition of the internal oxidizing group, C–N reductive elimination, and protonation. The oxidative addition of the internal oxidizing group changes the oxidation state of ruthenium from +2 to +4, while the C–N reductive elimination recovers the oxidation state of ruthenium from +4 to +2.The preference for the [4 + 1] annulation over the [4 + 2] annulation is attributed to the special structures of propargyl alcohols, because the existence of a β-H group and a hydroxyl group in alkynes is necessary for β-H elimination and enol–keto tautomerism. The use of alkynes without a β-H group or a hydroxyl group results in the formation of a [4 + 2] product.
The internal oxidizing group plays a key role in lowering the barrier height of C–N reductive elimination, since direct C–N reductive elimination from RuII is kinetically inaccessible. Hence, the [4 + 1] annulation is mediated by RuII/IV/II, while the [4 + 2] annulation is mediated by RuII/0/II.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21773010) and the Fundamental Research Funds for Tianjin Colleges (2018KJ171).Notes and references
- (a) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS PubMed; (b) M. Gulías and J. L. Mascarenas, Angew. Chem., Int. Ed., 2016, 55, 11000–11019 CrossRef PubMed; (c) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (d) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923–8930 CrossRef CAS PubMed; (e) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed; (f) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565–10569 CrossRef CAS PubMed; (g) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- (a) T. Zhou, L. B. Li, B. Li, H. B. Song and B. Q. Wang, Organometallics, 2018, 37, 476–481 CrossRef CAS; (b) Y. Yamamoto, K. Nishimura, S. Mori and M. Shibuya, Angew. Chem., Int. Ed., 2017, 56, 5494–5497 CrossRef CAS PubMed; (c) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (d) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed; (e) G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033 CrossRef CAS PubMed.
- (a) T. Fukutani, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 5198–5201 CrossRef CAS PubMed; (b) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) T. Fukutani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 2867–2874 CrossRef CAS PubMed.
- (a) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC; (b) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977–1979 CrossRef CAS PubMed; (c) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed; (d) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed; (e) J. L. Wu, X. L. Cui, L. M. Chen, G. J. Jiang and Y. J. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef CAS PubMed; (f) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed; (g) K. H. Ng, A. S. C. Chan and W. Y. Yu, J. Am. Chem. Soc., 2010, 132, 12862 CrossRef CAS PubMed; (h) B. Li, J. F. Ma, N. C. Wang, H. L. Feng, S. S. Xu and B. Q. Wang, Org. Lett., 2012, 14, 736–739 CrossRef CAS PubMed; (i) Z. Wang, P. Xie and Y. Xia, Chin. Chem. Lett., 2018, 29, 47 CrossRef CAS.
- (a) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 24 Search PubMed; (b) Z. Zhou, G. X. Liu and X. Y. Lu, Org. Lett., 2016, 18, 5668–5671 CrossRef CAS PubMed; (c) H. Huang, S. Nakanowatari and L. Ackermann, Org. Lett., 2017, 19, 4620–4623 CrossRef CAS PubMed; (d) S. Nakanowatari and L. Ackermann, Chem. – Eur. J., 2015, 21, 16246–16251 CrossRef CAS PubMed; (e) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- (a) K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2016, 138, 15527–15530 CrossRef CAS PubMed; (b) R. T. Yu, E. E. Lee, G. Malik and T. Rovis, J. Am. Chem. Soc., 2016, 128, 2782–2783 CrossRef PubMed; (c) A. Nishimura, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2012, 134, 15692–15695 CrossRef CAS PubMed; (d) K. Aikawa, Y. Hioki, N. Shimizu and K. Mikami, J. Am. Chem. Soc., 2011, 133, 20092–20095 CrossRef CAS PubMed.
- (a) D. R. Stuart, M. Bertrand-Laperle, K. M. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed; (b) J. D. Dooley, S. Reddy Chidipudi and H. W. Lam, J. Am. Chem. Soc., 2013, 135, 10829–10836 CrossRef CAS PubMed; (c) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625–16631 CrossRef CAS PubMed; (d) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426–12429 CrossRef CAS PubMed; (e) A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 7607–7610 CrossRef CAS PubMed; (f) A. Lerchen, S. Vásquez-Céspedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208–3211 CrossRef CAS PubMed.
- (a) L. Ackermann, J. Pospech, K. Graczyk and K. Rauch, Org. Lett., 2012, 14, 930 CrossRef CAS PubMed; (b) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS PubMed; (c) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., 2011, 123, 6503–6506 CrossRef; (d) S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef CAS PubMed; (e) B. Li, H.-L. Feng, S.-S. Xu and B.-Q. Wang, Chem. – Eur. J., 2011, 17, 12573–12577 CrossRef CAS PubMed; (f) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548–6551 CrossRef CAS PubMed; (g) C. Kornhaaß, C. Kuper and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1619–1624 CrossRef.
- (a) P. Liu, L. E. Sirois, P. H. Y. Cheong, Z.-X. Yu, I. V. Hartung, H. Rieck, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 10127–10135 CrossRef CAS PubMed; (b) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS PubMed; (c) H. Pellissier, Adv. Synth. Catal., 2018, 360, 1551–1583 CrossRef CAS; (d) Z.-J. Zuo, J. J. Liu, J. Nan, L. X. Fan, W. Sun, Y. Y. Wang and X. J. Luan, Angew. Chem., Int. Ed., 2015, 54, 15385–15389 CrossRef CAS PubMed; (e) R. Zeng and G.-B. Dong, J. Am. Chem. Soc., 2015, 137, 1408–1411 CrossRef CAS PubMed; (f) X. Hong, B. M. Trost and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 6588–6600 CrossRef CAS PubMed.
- (a) X. W. Wu, B. Wang, S. B. Zhou, Y. Zhou and H. Liu, Org. Lett., 2017, 19, 1294–1297 CrossRef CAS PubMed; (b) C. Q. Wang, L. Ye, C. Feng and T. P. Loh, J. Am. Chem. Soc., 2017, 139, 1762–1765 CrossRef CAS PubMed; (c) X. W. Wu and H.-T. Ji, J. Org. Chem., 2018, 83, 4650–4656 CrossRef CAS PubMed.
- X. W. Wu, B. Wang, S.-B. Zhou, Y. Zhou and H. Liu, ACS Catal., 2017, 7, 2494–2499 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) F. E. Jorge, A. Canal Neto, G. G. Camiletti and S. F. Machado, J. Chem. Phys., 2009, 130, 064108 CrossRef CAS PubMed; (b) C. T. Campos and F. E. Jorge, Mol. Phys., 2013, 111, 167 CrossRef CAS.
- (a) S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS; (b) D. Jacquemin, E. A. Perpete, G. Scalmani, I. Ciofini, C. Peltier and C. Adamo, Chem. Phys., 2010, 372, 61–66 CrossRef CAS.
- (a) J. Y. Tao, W. H. Mu, G. A. Chass, T. H. Tang and D. C. Fang, Int. J. Quantum Chem., 2013, 113, 975–984 CrossRef CAS; (b) D. C. Fang, SCRFRADII, Beijing Normal University, Beijing, China. Free of charge for academic users Search PubMed.
- (a) S. J. Grimme, Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed; (b) S. J. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. C. Fang, THERMO Program, Beijing Normal University, Beijing, China. Free of charge for academic users Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Universite de Sherbrooke, 2009; ( http://www.cylview.org) Search PubMed.
- (a) M. Zhang and G. Huang, Chem. – Eur. J., 2016, 22, 9356 CrossRef CAS PubMed; (b) J. L. Yu, S. Q. Zhang and X. Hong, J. Am. Chem. Soc., 2017, 139, 7224–7243 CrossRef CAS PubMed; (c) S. Ruiz, P. Villuendas, M. A. Ortuño, A. Lledós and E. P. Urriolabeitia, Chem. – Eur. J., 2015, 21, 8626 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- (a) N. Y. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929 CrossRef CAS PubMed; (b) A. Bechtoldt, C. Tirler, K. Raghuvanshi, S. Warratz, C. Kornhaass and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 264 CrossRef CAS PubMed.
- (a) V. Cadierno, S. E. García-Garrido, J. Gimeno, A. Varela-Álvarez and J. A. Sordo, J. Am. Chem. Soc., 2006, 128, 1360–1370 CrossRef CAS PubMed; (b) B. M. Trost, F. D. Toste and H. Shen, J. Am. Chem. Soc., 2000, 122, 2379–2380 CrossRef CAS.
- (a) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part A Structure and Mechanisms, Plenum Press, New York, 2nd edn, 1984 Search PubMed; (b) J. I. Seeman, Chem. Rev., 1983, 83, 83–134 CrossRef CAS.
- P. Atkins and J. D. Paula, Atkins’ Physical Chemistry, Oxford University Press, New York, 2014 Search PubMed.
- (a) P. Chen, Y. Sun, Y. Wu, L. Liu, J. Zhu and Y. Zhao, Org. Chem. Front., 2017, 4, 1482 RSC; (b) L. Liu, H. Yuan, T. Fu, T. Wang, X. Gao, Z. Zeng, J. Zhu, Y. Zhao and J. Org, Chem., 2014, 79, 80 CAS.
- (a) L. Xu, Q. Zhu, G. Huang, B. Cheng and Y. Xia, J. Org. Chem., 2012, 77, 3017–3024 CrossRef CAS PubMed; (b) B. Lian, L. Zhang, S. J. Li, L. L. Zhang and D. C. Fang, J. Org. Chem., 2018, 83, 3142–3148 CrossRef CAS PubMed; (c) L. Zhang and D. C. Fang, Organometallics, 2017, 36, 4943–4951 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo00154a |
| This journal is © the Partner Organisations 2019 |