
Cross coupling of alkylsilicates with acyl chlorides via photoredox/nickel dual catalysis: a new synthesis method for ketones†
Cyril
Ollivier  *a
and
Louis
Fensterbank
*a
*a
and
Louis
Fensterbank
*a
*a
and
Louis
Fensterbank
*a
Abstract
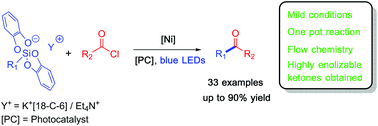
Photoredox/nickel dual catalysis using easily oxidized bis-catecholato hypercoordinated silicon derivatives as radical sources and acyl chlorides as electrophiles allows a new method of formation of dialkyl and alkyl-aryl ketones as well as dibenzyl ketones which are less easily accessed. Flow chemistry can be used.
 Please wait while we load your content...
Please wait while we load your content...

