
Genome mining for fungal polyketide-diterpenoid hybrids: discovery of key terpene cyclases and multifunctional P450s for structural diversification†
 *a
Yudai
Matsuda
*f
*a
Yudai
Matsuda
*f
Abstract
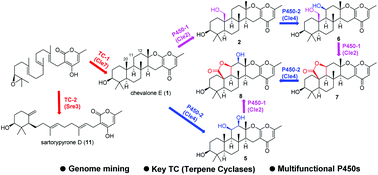
A biosynthetic gene cluster for chevalone E (1) and its oxidized derivatives have been identified within the genome of the endophytic fungus Aspergillus versicolor 0312, by a mining strategy targeting a polyketide-diterpenoid hybrid molecule. The biosynthetic pathway has been successfully reconstituted in the heterologous fungus Aspergillus oryzae. Interestingly, two P450 monooxygenases, Cle2 and Cle4, were found to transform 1 into seven new analogues including 7 and 8 that possess a unique five-membered lactone ring. Furthermore, the replacement of the terpene cyclase gene with that from another fungus led to the production of sartorypyrone D (11), which has a monocyclic terpenoid moiety. Finally, some of the compounds obtained in this study synergistically enhanced the cytotoxicity of doxorubicin (DOX) in breast cancer cells.
 Please wait while we load your content...
Please wait while we load your content...

