
Synthesis of azulenophthalimides by phosphine-mediated annulation of 1,2-diformylazulenes with maleimides†

Abstract
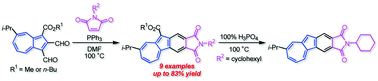
The reaction of 1,2-diformylazulenes with several maleimides in the presence of PPh3 gave the corresponding azulenophthalimides with an ester function at the five-membered ring in moderate to good yields. The removal of the ester function from the azulenophthalimide with an N-cyclohexyl function was also achieved by the treatment with 100% H3PO4. The optical and electrochemical properties of the azulenophthalimides were investigated by UV/Vis measurements, voltammetry experiments and DFT calculations. The azulenophthalimide without the ester function exhibited remarkable emission under acidic conditions. Moreover, the azulenophthalimides displayed a significant spectral change under electrochemical reduction conditions.
 Please wait while we load your content...
Please wait while we load your content...

