
Efficient and phosphine-free bidentate N-heterocyclic carbene/ruthenium catalytic systems for the dehydrogenative amidation of alcohols and amines†
 *a
and
Francis
Verpoort
*aef
*a
and
Francis
Verpoort
*aef
Abstract
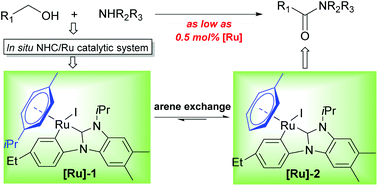
The direct amide synthesis from alcohols and amines applying various transition metal catalysts has been demonstrated as an attractive and promising process. Among various catalytic systems, N-heterocyclic carbene (NHC)-based ruthenium (Ru) ones have been testified to be active for this atom-economic transformation. Although a variety of imidazole-based NHC/Ru catalytic systems were reported to be active for this reaction, the benzimidazole-based analogs exhibited higher catalytic performance in most cases. However, these catalytic systems, which comprise a monodentate NHC ligand and a Cl or phosphine ligand as the key components, require relatively high catalyst loadings. In order to obtain more active and robust catalytic systems, we aim to bridge two monodentate ligands with one bidentate NHC ligand. Therefore, a number of CNHCC bidentate NHC precursors were designed and synthesized. Through screening of the NHC precursors and other reaction conditions, potent and phosphine-free bidentate NHC/Ru catalytic systems were discovered for the efficient amide synthesis. Interestingly, from the in situ generated catalytic system, two NHC/Ru intermediates were isolated and structurally confirmed by X-ray crystallography. Notably, these two complexes are active for the amide synthesis even at a low catalyst loading of 0.5 mol%, which could verify that they should be key intermediates during the catalysis. Probably, the current catalytic systems, featuring high efficiency and ready accessibility, could be valuable for more interesting applications.
 Please wait while we load your content...
Please wait while we load your content...

