
Nickel catalyzed synthesis of 4,4′-bichromenes/4,4′-bithiochromenes and their Atropisomerism†
Somasundaram
Muthuramalingam‡
a,
Jai Anand
Garg‡
b,
R.
Karthick
c,
Thomas
Fox
d,
Olivier
Blacque
d,
Koushik
Venkatesan
 de,
Saiganesh
Ramanathan
c,
Senthamaraikannan
Kabilan
*a and
K. K.
Balasubramanian
*f
de,
Saiganesh
Ramanathan
c,
Senthamaraikannan
Kabilan
*a and
K. K.
Balasubramanian
*f
aDepartment of Chemistry, Annamalai University, Annamalainagar-608002, India. E-mail: profdrskabilanau@gmail.com
bDepartment of Chemistry, Indian Institute of Technology-Madras, Adyar, Chennai-600036, India
cStrides-Shasun pharmaceuticals Ltd, Vandalur-Kelambakkan Rd., Chennai-600127, India
dDepartment of Chemistry, University of Zürich, Zürich, CH-8057, Switzerland
eDepartment of Molecular Sciences, Macquarie University, NSW 2109, Australia
fDepartment of Biotechnology, Indian Institute of Technology-Madras. Adyar, Chennai-600036, India
First published on 22nd November 2018
Abstract
Axially chiral molecules have established undisputed importance in both medicinal chemistry and enantiomeric catalysis. The size, shape and hybridization of the substituent adjacent to the rotational axis greatly dictate atropisomerism. Herein, we examined the tropoisomeric behavior of 4,4′-bichromenes and 4,4′-bithiochromenes derivatives that were synthesized by a nickel catalyzed reductive homocoupling strategy. The as-synthesized 3,3′-disubstituted 4,4′-bichromenes displayed atropisomerism, as evidenced by chiral stationary phase HPLC, electronic circular dichroism and single crystal XRD studies. Insights into the torsional barrier about the internuclear axis in 4,4′-bichromenes were gained both experimentally and theoretically (DFT studies). The lower activation energy barrier (Ea) of approximately 12 kcal mol−1 as compared to that of fully aromatic 1,1′-binapthyl explains the conformationally unstable nature of 3,3′-unsubstituted 4,4′-bichromenes at room temperature.
Introduction
The atroposelective synthesis of axially chiral biaryls is considered significant since it can afford a variety of biologically active molecules and excellent chirality transfer agents. Molecules such as Vancomycin and Diphosphine BINAP bear testimony to this fact.1–9 An important factor in the synthesis of dissymmetric biaryls is the presence of a substituent adjacent to the internuclear rotational axis. The steric size, shape and hybridization of these substituents exert a large influence on the optical stability of the molecules.10 While atropisomerism in 1,1′-binaphthyls 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 has been largely studied, the closely related condensed heterocyclic systems have received less attention. Among a few of the heterocyclic analogues examined, namely, 4,4′-biquinolines 2, 4,4′-bi-2,2′-quinolones 3, and 1,1′-biisoquinolines 4,12,13 both 2 and 3 (when R = H) have been optically resolved and found to be conformationally stable at ambient temperatures, similar to 1,1′-binaphthyl (Fig. 1).6c,13,14 In contrast, it is interesting to note that 1,1′-biisoquinoline 4 exhibits rapid racemization due to the low transannular steric hindrance between H-8(8′) and N-2(2′).15 Mazzanti and Schlosser have also shown that the stereogenic torsional barrier of arylpyridines is up to 4.2 kcal mol−1 smaller than that of biphenyl due to the compressibility of the nitrogen lone pair.16 These facts provoked our interest to examine atropisomerism in 4,4′-bichromenes 6 and 4,4′-bithiochromenes 7, which are a medicinally important class of compounds closely related to 4,4′-bicoumarins 5.6a,17 Moreover, the synthetic chemistry of 4,4′-bichromenes has been scarcely explored and only a few reports exist.6b,18 In this paper, we present the first report on the atropisomerism of 3,3′-disubstituted-4,4′-bichromenes 6, and 3,3′-disubstituted-4,4′-bithiochromenes 7. For comparative studies we have also prepared a few partially aromatic biaryls, namely, 3,3′,4,4′-tetrahydro-1,1′-binapthyls (8a and 8b), and studied their axial chirality (see chart S1.0 in the ESI†).
11 has been largely studied, the closely related condensed heterocyclic systems have received less attention. Among a few of the heterocyclic analogues examined, namely, 4,4′-biquinolines 2, 4,4′-bi-2,2′-quinolones 3, and 1,1′-biisoquinolines 4,12,13 both 2 and 3 (when R = H) have been optically resolved and found to be conformationally stable at ambient temperatures, similar to 1,1′-binaphthyl (Fig. 1).6c,13,14 In contrast, it is interesting to note that 1,1′-biisoquinoline 4 exhibits rapid racemization due to the low transannular steric hindrance between H-8(8′) and N-2(2′).15 Mazzanti and Schlosser have also shown that the stereogenic torsional barrier of arylpyridines is up to 4.2 kcal mol−1 smaller than that of biphenyl due to the compressibility of the nitrogen lone pair.16 These facts provoked our interest to examine atropisomerism in 4,4′-bichromenes 6 and 4,4′-bithiochromenes 7, which are a medicinally important class of compounds closely related to 4,4′-bicoumarins 5.6a,17 Moreover, the synthetic chemistry of 4,4′-bichromenes has been scarcely explored and only a few reports exist.6b,18 In this paper, we present the first report on the atropisomerism of 3,3′-disubstituted-4,4′-bichromenes 6, and 3,3′-disubstituted-4,4′-bithiochromenes 7. For comparative studies we have also prepared a few partially aromatic biaryls, namely, 3,3′,4,4′-tetrahydro-1,1′-binapthyls (8a and 8b), and studied their axial chirality (see chart S1.0 in the ESI†).
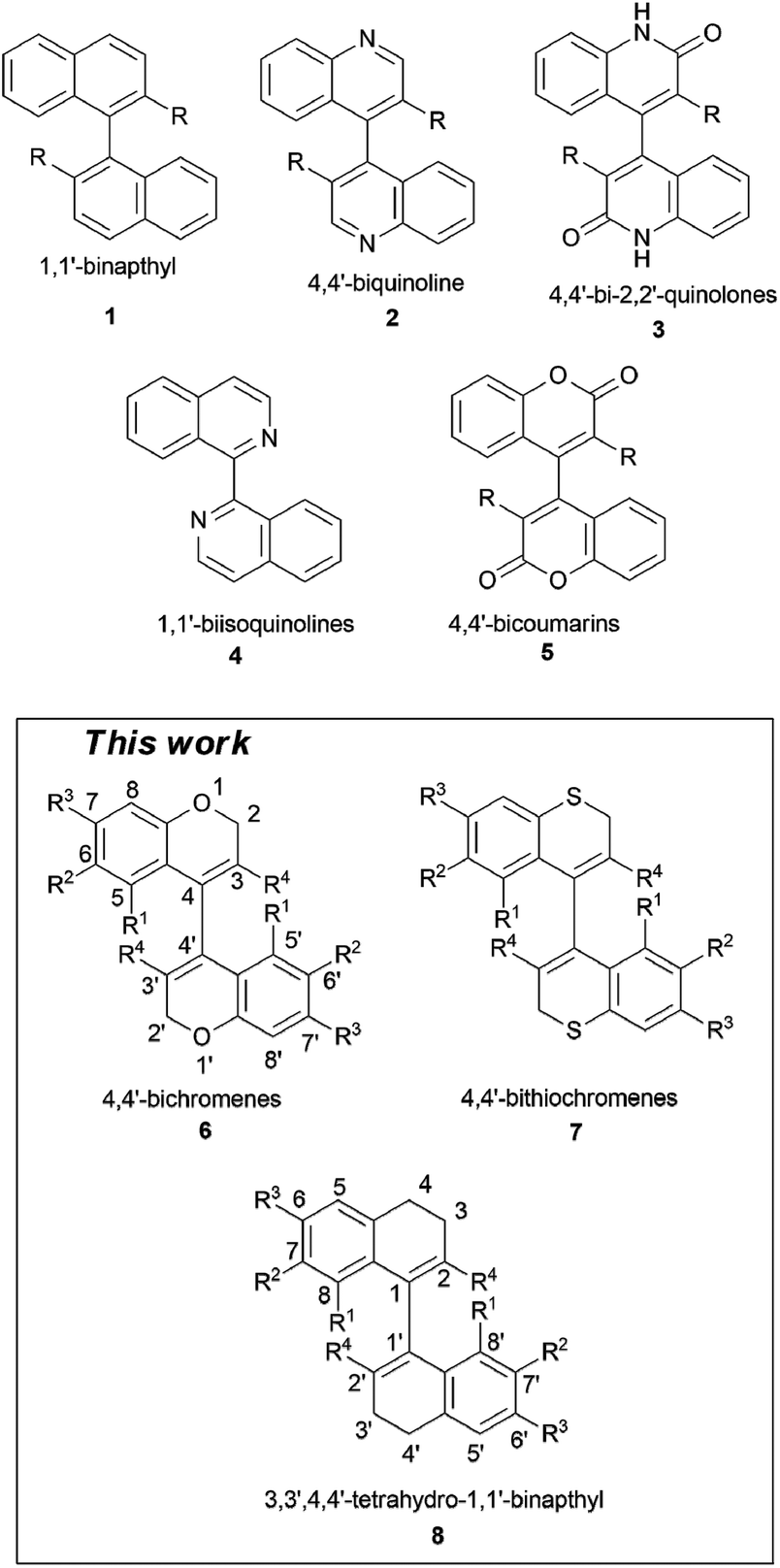 | ||
| Fig. 1 Examples of various classes of biaryl systems. | ||
Results and discussion
We began our investigation with the synthesis and examination of atropisomerism in 7,7′-dimethyl-4,4′-bichromene 6f (Fig. 2).19 The chiral HPLC trace of 6f showed only a single eluting peak. In the 1H NMR spectrum, the –OCH2 protons appeared as a doublet at 4.84 ppm and the olefinic proton appeared as a triplet at 5.74 ppm. The non-diastereotopic nature of the methylene signals indicated unresolvable fast rotation about the stereogenic axis at room temperature (ESI, S3.6 and S4.3†). | ||
| Fig. 2 Various biaryl compounds preliminarily examined for axial chirality at room temperature. | ||
The above observations were unlike those of 1,1′-binaphthyl 111a or 4,4′-biquinoline 2,15 which exhibited axial chirality at room temperature. Furthermore, we also prepared the tetrahydro analogue of 1, namely, the 3,3′,4,4′-tetrahydro-1,1′-binaphthalene20 (8) (R1 = R2 = R3 = R4 = H) (Fig. 1) and analysed its conformational stability. It too displayed an achiral behaviour at room temperature as revealed by 1H NMR studies and HPLC analysis performed with six different chiral stationary phase columns (ESI, S3.15 and S4.10†). Finally, in line with the general expectation, we sought to improve the optical stability by functionally substituting 3,3′-positions in the bichromene/bithiochromene systems. Accordingly, a preliminary examination of the room temperature 1H NMR spectrum of 3,3′-dibromo-2H,2′H-4,4′-bichromene (12) indeed revealed the expected diastereotopic pattern (ESI, S3.18†).21 Recently, Lee et al.22 have reported an iodine monochloride (ICl)-mediated synthesis of substituted 3,3′-diiodo-2H,2′H-4,4′-bichromene from diynes. The 1H NMR spectrum of the diiodo compound also suggested atropisomerism similar to our compounds (Fig. 2). Next, we aimed to develop an easier protocol for synthesizing 3,3′-disubstituted 4,4′-bichromenes, which would provide direct access to these heterocyclic axially chiral entities. The metal-mediated homocoupling of 4-chloro-3-formylchromenes (9) appeared to be the most expedient route to access diversely functionalized potentially chiral 4,4′-bichromenes. β-Chlorovinyl aldehydes are versatile synthons and their chemistry has been recently reviewed.23–26
Though there exists few reports on the metal-catalyzed homocoupling and cross coupling of β-chlorovinyl aldehydes with acetylenes,26 arylboronic acids and aryl amines,27,28 surprisingly no reports were found for the metal mediated homocoupling of 4-chloro 3-formylchromenes. Homocoupling of aryl halides can be accomplished using various metal-catalysed systems. Our initial attempts to realize the homocoupling of 4-chloro-3-formylchromenes (9) using CuI/Cu/Ni(PPh3)2Cl2 or Cu/Pd(PPh3)4 or indium/Ni(PPh3)2Cl2 in different solvents and conditions were unsuccessful.28 Finally, we succeeded in the synthesis of the hitherto unknown 4,4′-bichromene-3.3′-dicarboxaldehyde 6a in 70% yield by homocoupling chloroaldehyde 9a using an Ni(0) complex generated in situ by the reduction of Ni(PPh3)2Cl2 with activated zinc29 (Scheme 1). 4,4′-Bichromene-3,3′-dicarboxaldehyde 6a, isolated as a crystalline solid, was thoroughly characterized by 1H NMR,13C NMR spectroscopy and HRMS. A cursory glance of its 1H NMR spectrum revealed this molecule to be chiral (ESI, S3.1†). The 1H NMR spectrum showed an AB quartet for the –OCH2 (J = 14.5 Hz) protons, revealing their diastereotopic nature in contrast to those of the simple 4,4′-bichromene and 4,4′-bithiochromene (6f and 7).
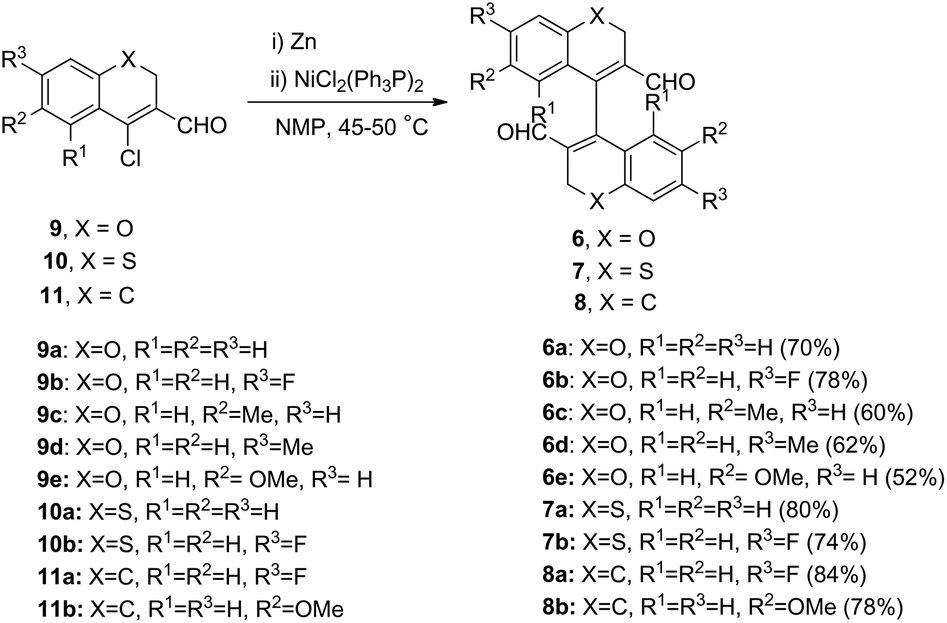 | ||
| Scheme 1 NiCl2(PPh3)2 and Zn catalyzed reductive homocoupling of chloroaldehydes. | ||
The chiral HPLC traces for 6b showed two peaks in 1:1 ratio, further confirming it to be a racemate (ESI S4.11 and Fig. S1†). The racemate, 6b, was separated into optical antipodes by chiral preparative HPLC. The chiral purity of the separated antipodes was rechecked by chiral HPLC analysis. The electronic circular dichroism (CD) spectrum for both enantiomers were measured and determined to be mirror images, displaying the cotton effect at 300 nm and 400 nm (Fig. 3, and ESI, S5.0†).
 | ||
| Fig. 3 The electronic circular dichroism spectrum of 6b. | ||
Single crystals of 6a, suitable for XRD analysis, were obtained from slow evaporation of the compound in methanol at room temperature (ESI, S6.0†). The crystals obtained were in the racemic form. In line with our expectations, the two bichromene units were flanked along the internuclear axis in a non-coplanar fashion. The dihedral angle between the mean molecular aryl ring planes was found to be 88.29°, indicating a close perpendicular conformation. The distance of the internuclear axial bond, viz., C4–C4′ was found to be 1.5009(19) Å, which is close in comparison to the C1–C1′ distance in (R)-(−)-1,1′-binaphthyl- (1.511(7) Å).30a We also obtained racemic single crystals of compound 8; the molecular structure revealed a cisoid configuration with mean planes for the aryl rings subtending an angle of 66.11° about the stereogenic axis (Fig. 4).
 | ||
| Fig. 4 X-ray crystallographic structure of 6a (left) and 8 (right). | ||
The internuclear distance for C1–C11 was found to be 1.477(6) Å. These distances and angles were quite similar to that of racemic 1,1′-binapthyl crystal,30b which showed an internuclear distance of 1.475 Å and an angle of 68° between the planes of the binapthyl residues. Using the facile homocoupling methodology described above, we synthesized for the first time 4,4′-bichromenes-3.3′-dicarboxaldehydes 6a–e, 4,4′-bithiochromenes-3.3′-dicarboxaldehydes 7a–b, and 3,3′,4,4′-tetrahydro-[1,1′-binaphthalene]-2,2′-dicarbaldehydes 8a–b (Scheme 1). All these compounds (6a–e, 7a–b, and 8a–b) showed atropisomerism, as revealed by their 1H NMR spectra. Furthermore, isomeric resolution of peaks was observed on chiral HPLC traces of 6b, 6e–j, 7a, and 8a (ESI, S4.0†). Moreover, by simple functional group transformation of 6b and 6a, a few more novel 3,3′-disubstituted-4,4′-bichromenes, viz., 6g–6i and 6j, respectively were obtained (Scheme 2 and Experimental section). Their chiral behaviour was also observed with 1H NMR and chiral HPLC analyses. The acid 6g, alcohol 6h and the oxime 6j displayed atropisomerism. HPLC analysis of the cyano derivative 6i showed two peaks in 1:1 ratio, revealing it to be conformationally stable. However, the 1H NMR spectrum exhibited only a singlet and no an AB quartet for the –OCH2 protons. This is possibly due to accidental equivalence (ESI, S3.9†). We are currently investigating the direct enantioselective synthesis of 3,3′-disubstituted 4,4′-bichromenes by asymmetric catalysis.
 | ||
| Scheme 2 Conversion of 4,4′-bichromenes-3.3′-dicarboxaldehydes to other functionalities viz. acid, alcohol, nitrile and oximes. | ||
As mentioned earlier, the reason for the higher degree of rotational freedom in the case of 4,4′-bichromenes was due to the reduction in transannular steric hindrance about the internuclear axis. However, the shape and hybridization factor of the substituent atoms, which presumably influences this effect, has neither been experimentally investigated nor empirically suggested in the system under consideration. In order to gain more insight into the conformational behaviour of the 3,3′-unsubstituted 4,4′-bichromene systems, the two compounds 6k and 6l (Fig. 2 and ESI, S2.0†) were synthesized and collected for further investigation. The coalescence temperature of the exchanging isomers was determined from the variable temperature dynamic 1H NMR studies31 for the sample in deuterated THF. The rate constant (Kr) thus determined was used for calculating activation energies (Ea). The activation parameters (ΔH‡ and ΔS‡) were also obtained employing the Eyring equation (ESI, S7.0†). The activation energies (Ea) of 6k and 6l were found to be 14.6 kcal mol−1 and 11.16 kcal mol−1, respectively. The rotational energy barrier was reflected in the Gibbs free energy (ΔG‡) difference between the ground state and the rate-limiting transition state. The estimated values of ΔG‡ for 6k and 6l were 13.44 kcal mol−1 and 10.75 kcal mol−1, respectively. These values were approximately half of the experimentally determined value of ΔG‡ for carba-analogous 1,1′-binapthyl (23.5 kcal mol−1) in DMF at 50 °C.32a
DFT calculations were also performed to determine the activation energy Ea for free rotation (ESI, S8.0†). The Ea of 6k and 6l generated by DFT computation were close values of 19.15 kcal mol−1 and 20.66 kcal mol−1, respectively. A table of comparison is provided in the ESI (Table S1†). As expected, the Ea for 4,4′-bichromenes 6k and 6l was lower than the Ea for the 1,1′-binapthyl system by approximately 12 kcal mol−1. Comparing the difference in the activation energies for bichromene versus 1,1′-binapthyl, both experimental and DFT computed values agreed on the lower value of ∼10–13 kcal mol−1.32 In general, the DFT results overestimated the activation energies by approximately 5–8 kcal mol−1, but were consistent with the trend. The marginal difference observed in the experimentally derived values of (Ea) and ΔH‡ for 6k and 6l is also reflected in the DFT calculations, indicating the limited influence from non-sterically interfering dimethyl substituents at the 2,2′-positions. We also calculated the rotational barrier of a hypothetical 4,4′-bichromene with a methyl substituent at the 3,3′-position, namely, 3,3′-dimethyl-4,4′-bichromene was calculated and compared to with that of 2,2′-dimethyl-1,1′-binapthyl (ESI, S8†). The rotational barrier of the former was calculated to be 80 kcal mol−1 and the later to be 110 kcal mol−1. The sufficiently high energy barrier of 80 kcal mol−1 suggests the compound to be atropisomeric at RT, though not to the extent of its aromatic counterpart. This computation suggests that even smaller sterically hindering substituents such as the methyl group at the 3,3′-position of bichromene could facilitate atropisomerism.
From the above studies, it is possible to surmise a few reasons for the difference in atropisomerism between the binapthyl and bichromene systems. (i) The non-planarity of the heterocyclic ring in the bichromene system (presence of sp3 carbon) can cause lower aromatic stabilization of either the syn- or anti isomers, therefore decreasing the energy barrier for rotation. In contrast, the sp2 carbon bearing the fully aromatic binapthyl system was more stable due to conjugation effects. (ii) The possibility of any non-bonded repulsive interaction between the olefinic proton and the benzene ring in the bichromene/bithiochromene system was lower than that of the binapthyl system favouring free-rotation. This was also manifested in the conformationally unstable nature of 3,4,3′,4′-tetrahydro-1,1′-binapthalene 8 in contrast to that of 1,1-binapthyl 1.
Conclusions
In summary, we present the first report on atropisomerism in 4,4′-bichromenes and 4,4′-bithiochromenes. A nickel-catalyzed reductive homocoupling strategy was applied for the synthesis of several analogues of 4,4′-bichromenes and 4,4′-bithiochromenes. The configurational stability of a few 4,4′-bichromenes were studied experimentally and theoretically to be compared with the 1,1′-binapthyl system. We believe that this study will be helpful in designing novel heteroaromatic axially chiral biaryl motifs that are useful for enantioselective catalysis and in medicinal chemistry.Experimental section
Typical procedure for the Ni-catalysed reductive homocoupling reaction is as follows. To a stirred solution of 4-chloro-3-formylchromene (10.0 mmol) in freshly distilled NMP (10.0 mL), activated zinc powder (2.0 g, 30.0 mmol) and NiCl2(PPh3)2 (0.67 g, 1.0 mmol) were added at 45–50 °C. The reaction mixture was stirred at the same temperature for 3–4 h and the progress was monitored by TLC. After completion of the reaction, the reaction mass was cooled to room temperature and diluted with 20 mL of methyl tert-butyl ether (MTBE). The resulting solution was then filtered through a Celite bed and washed further with 10 mL of MTBE. The combined organic portions were washed with dilute 5% aqueous HCl (2 × 15 mL), followed by water (1 × 15 mL), dried over anhydrous sodium sulphate and then concentrated under reduced pressure to yield brown pasty solids. The crude product thus obtained was triturated with 8 mL of ethanol, filtered and dried to produce off-white to yellow solids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the following people: Head of the department of chemistry, IIT Madras for support with HRMS and low VT-NMR analysis; Mr V. Ramkumar for XRD analysis; Prof. G. Sekar, Ms Sangeetha and Mr Y. Chari of IIT Madras for HPLC analysis; Prof. Sivaguru and Dr Josepha Lourdaraj, North Dakota State University, USA for the chiral separation and CD analysis; Prof. Anju Chadha and Mr Kabilan. C., Department of Biotechnology, IIT Madras for help with HPLC analyses. Strides-Shasun Ltd, Chennai is acknowledged for analytical support and encouragement. K. K. B thanks INSA, New Delhi for the award of an INSA Senior Scientist position and J. A. G thanks SERB, India (Grant No. YSS/2015/001695) for funds.Notes and references
- (a) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS PubMed; (b) G. Bringman, M. Breuning and S. Tasler, Synthesis, 1999, 525 CrossRef; (c) G. Bringmann, M. Breuning, R. Walter, A. Wuzik, K. Peters and E.-M. Peters, Eur. J. Org. Chem., 1999, 3047 CrossRef CAS; (d) Z. Wu, C. Wang, L. N. Zakharov and P. R. Blakemore, Synthesis, 2014, 46, 678 CrossRef; (e) S. Yu and L. Pu, Tetrahedron, 2015, 71, 745 CrossRef CAS.
- B. H. Lipshutz and J. M. Keith, Angew. Chem., Int. Ed., 1999, 38, 3530 CrossRef CAS PubMed.
- (a) J. Seyden-Penne, in Chiral Auxiliaries and Ligands in Asymmetric Synthesis, Wiley, New York, 1995 Search PubMed; (b) Comprehensive Asymmetric Catalysis I-III, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999 Search PubMed; (c) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS PubMed; (d) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS; (e) P. Kočovský, S. Vyskočil and M. Smrčina, Chem. Rev., 2003, 103, 3213 CrossRef PubMed; (f) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC.
- (a) C. Zhu, Y. Shi, M.-H. Xu and G. Q. Lin, Org. Lett., 2008, 10, 1243 CrossRef CAS PubMed; (b) A. I. Meyers, T. D. Nelson, H. Moorlag, D. J. Rawson and A. Meier, Tetrahedron, 2004, 60, 4459 CrossRef CAS; (c) J. Vachon, C. Perollier, D. Monchaud, C. Marsol, K. Ditrich and J. Lacour, J. Org. Chem., 2005, 70, 5903 CrossRef CAS PubMed.
- (a) T. Yamamoto, T. Maruyama, Z. Zhou, T. Ito, T. Fukada, Y. Yoneda, F. Begum, T. Ikeda, S. Sasaki, H. Takezoe, A. Fukada and K. Kubota, J. Am. Chem. Soc., 1994, 116, 4832 CrossRef CAS; (b) Y. Chao, G. R. Weisman, G. D. Y. Sogah and D. J. Cram, J. Am. Chem. Soc., 1979, 101, 515 CrossRef.
- (a) J.-G. Lei, M.-H. Xu and G.-Q. Lin, Synlett, 2004, 2364 CAS; (b) J. D. Hepworth, T. K. Jones and R. Livingstone, Tetrahedron, 1981, 37, 2613 CrossRef CAS; (c) J. Hashim and C. O. Kappe, Adv. Synth. Catal., 2007, 349, 2353 CrossRef CAS.
- (a) C. Rosini, L. Franzini, A. Raffaelli and P. Salvadori, Synthesis, 1992, 503 CrossRef CAS; (b) J. K. Whitesell, Chem. Rev., 1989, 89, 1581 CrossRef CAS.
- L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS PubMed.
- (a) M. Oki, in The Chemistry of Rotational Isomers, Springer-Verlag, Berlin, 1993 CrossRef; (b) Y. Badar, A. S. Cooke and M. M. Harris, J. Chem. Soc., 1965, 1412 RSC; (c) A. S. Cooke and M. M. Harris, J. Chem. Soc. C, 1967, 988 RSC.
- (a) T. W. Wallace, Org. Biomol. Chem., 2006, 4, 3197 RSC; (b) O. Baudoin, Eur. J. Org. Chem., 2005, 4223 CrossRef CAS; (c) S. Reichert and B. Breit, Org. Lett., 2007, 9(5), 899 CrossRef CAS PubMed; S. Banerjee, B. E. Riggs, L. N. Zakharov and P. R. Blakemore, Synthesis, 2015, 47, 4008 Search PubMed.
- (a) L. Pu, in 1,1-Binaphthyl based Chiral Materials: Our Journey, Imperial College Press, 2009 CrossRef; (b) S. Singh, Curr. Sci., 1975, 44(24), 873 CAS; (c) T. Hoshi, E. Nozawa, M. Katano, T. Suzuki and H. Hagiwara, Tetrahedron Lett., 2004, 45, 3485 CrossRef CAS.
- S. Ray and Kr. P. Subal, J. Indian Chem. Soc., 2005, 82(3), 236 CAS.
- M. Slany and P. J. Stang, Synthesis, 1996, 1019 CrossRef CAS.
- M. Crawford and I. F. B. Smyth, J. Chem. Soc., 1954, 3464 RSC.
- T. Itakura, R. Tsuchiya, K. Kobayashi, H. Takahashi and K.-I. Hirao, J. Chem. Soc., Perkin Trans. 1, 1999, 3677 Search PubMed.
- A. Mazzanti, L. Lunazzi, S. Lepri, R. Ruzziconi and M. Schlosser, Eur. J. Org. Chem., 2011, 6725 CrossRef CAS.
- (a) M. IIyas and M. Parveen, Tetrahedron, 1996, 52, 3991 CrossRef; (b) K. Gnanaguru, N. Ramasubbu, K. Venkatesan and V. Ramamurthy, J. Org. Chem., 1985, 50, 2337 CrossRef CAS; (c) D. M. X. Donnelly, J. P. Finet, P. J. Guiry and K. Nesbitt, Tetrahedron, 2001, 57, 413 CrossRef CAS; (d) M. Rahman, M. Riaz and U. R. Desai, Chem. Biodiversity, 2007, 47, 2495 CrossRef PubMed.
- K. K. Balasubramanian, K. Virupakshareddy and R. Nagarajan, Tetrahedron Lett., 1973, 50, 5003 CrossRef.
- See ESI† for the synthetic procedure of 6f.
- E. I. Troyansky, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2001, pp. 1–6 Search PubMed.
- R. V. S. Kumar , unpublished results, Ph.D dissertation work, 2018. Note: The preliminary examination of 12 has been done by 1H NMR spectroscopy; the complete characterization will be published in a separate study.
- J. Mo, W. Choi, J. Min, C.-E. Kim, D. Eom, S. H. Kim and P. H. Lee, J. Org. Chem., 2013, 78, 11382 CrossRef CAS PubMed.
- For review see: S. Brahma and J. K. Ray, Tetrahedron, 2008, 64, 2883 CrossRef CAS.
- (a) P. Karthikeyan, M. A. Rani, R. Saiganesh, K. K. Balasubramanian and S. Kabilan, Tetrahedron, 2009, 65, 811 CrossRef CAS; (b) K. Perumal, J. A. Garg, O. Blacque, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Chem. – Asian J., 2012, 7, 2670 CrossRef CAS PubMed; (c) M. Koch, K. Perumal, O. Blacque, J. A. Garg, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Angew. Chem., Int. Ed., 2014, 53, 6378 CrossRef CAS PubMed.
- S. Hesse and G. Kirsch, Synthesis, 2001, 755 CrossRef CAS.
- S. Hesse and G. Kirsch, Tetrahedron, 2005, 61, 6534 CrossRef CAS.
- S. Some, J. K. Ray, M. G. Bunwell and M. T. Jones, Tetrahedron Lett., 2007, 48, 3609 CrossRef CAS.
- (a) S. Some, B. Dutta and J. K. Ray, Tetrahedron Lett., 2006, 47, 1221 CrossRef CAS; (b) G. Bringmann, R. Walter and R. Weirich, Angew. Chem., Int. Ed. Engl., 1990, 29, 977 CrossRef.
- (a) W.-W. Chen, Q. Zhao, M.-H. Xu and G.-Q. Lin, Org. Lett., 2010, 12, 1072 CrossRef CAS PubMed; (b) M. Iyoda, H. Otsuka, K. Sato, N. Nisato and M. Oda, Bull. Chem. Soc. Jpn., 1990, 63, 80 CrossRef CAS.
- (a) R. Kuroda and S. F. Mason, J. Chem. Soc., Perkin Trans. 2, 1981, 167 RSC; (b) K. A. Kerr and J. M. Robertson, J. Chem. Soc. B, 1969, 1146 RSC.
- (a) D. Kost, E. H. Carlson and M. Raban, J. Chem. Soc. D, 1971, 656 RSC; (b) H. S. Gutowsky and C. H. Holm, J. Chem. Phys., 1956, 25, 1228 CrossRef CAS; (c) J. Heidberg, J. A. Weil, G. A. Janusonis and J. K. Anderson, J. Chem. Phys., 1964, 41, 1033 CrossRef CAS.
- (a) A. S. Cooke and M. M. Harris, J. Chem. Soc., 1963, 2365 RSC; (b) A. K. Colter and L. M. Clemens, J. Phys. Chem., 1964, 68, 651 CrossRef CAS; (c) L. Meca, D. Rÿeha and Z. Havlas, J. Org. Chem., 2003, 68, 5677 CrossRef CAS PubMed; (d) E. P. Kyba, G. W. Gokel, F. de Jong, K. Koga, L. R. Sousa, M. G. Siegel, L. Kaplan, D. Y. Sogah and D. J. Cram, J. Org. Chem., 1977, 42, 4173 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR HPLC, single crystal XRD data and computational details are provided. CCDC 8516921817591. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00820e |
| ‡ These authors contributed equally to the work. |
| This journal is © the Partner Organisations 2019 |