
Incorporation of narcissistic self-sorting supramolecular interactions for the spontaneous fabrication of multiple-color solid-state materials for OLED applications†‡
Yu-Tang
Tsai
a,
Guillaume
Raffy
 a,
Hsiang-Fang
Liu
b,
Bo-Ji
Peng
b,
Kuo-Pi
Tseng
b,
Lionel
Hirsch
c,
André
Del Guerzo
a,
Dario M.
Bassani
*a and
Ken-Tsung
Wong
*bd
a,
Hsiang-Fang
Liu
b,
Bo-Ji
Peng
b,
Kuo-Pi
Tseng
b,
Lionel
Hirsch
c,
André
Del Guerzo
a,
Dario M.
Bassani
*a and
Ken-Tsung
Wong
*bd
aUniv. Bordeaux, CNRS, Bordeaux INP, ISM,UMR 5255, F-33400, Talence, France. E-mail: dario.bassani@u-bordeaux.fr
bDepartment of Chemistry, National Taiwan University, Taipei, 10617, Taiwan. E-mail: kenwong@ntu.edu.tw
cIMS, Univ. Bordeaux, Bordeaux INP, ENSCBP, CNRS UMR 5218, F-33400 Talence, France
dInstitute of Atomic and Molecular Science, Academia Sinica, Taipei, 10617, Taiwan
First published on 21st November 2019
Abstract
A series of six compounds composed of an emissive oligofluorene or dithiophenethiobenzimidazole (p3) core bearing H-bonding biuret molecular recognition motifs in the meta- or para-position was investigated with regard to their propensity to undergo either social or narcissistic self-sorting when deposited from dilute THF solutions. The structural similarity of the compounds uniformly leads to social self-sorting with the exception of two combinations involving p3 for which narcissistic self-sorting is observed instead. Their behavior is attributed to the difference in location of the biuret groups (meta vs. para) combined with a greater structural difference in the conjugated core. Deposition of solutions demonstrating narcissistic self sorting gives rise to ensembles of disk-like aggregates exhibiting two different populations of emission (blue and magenta or yellow and orange).
Introduction
Self-sorting is defined as the ability of a multi-component system to undergo spontaneous organization based on a specific property or behavior.1 It is ubiquitous in living organisms, where it is present at the cellular, sub-cellular, and molecular levels, as well as manifesting itself in the social behaviour of individual organisms.2 At the molecular scale, self-sorting may be termed narcissistic or social, depending on whether the final outcome results in the grouping of typologically similar (narcissistic self-sorting) or dissimilar molecules (social self-sorting).3–5 This concept has proven to be particularly useful in understanding and predicting the assembly of molecular sub-components into mixtures of well-defined supramolecular architectures and, in this regard, it bridges the gap between molecular self-assembly and more complex, evolutionary systems based on multi-component libraries.6,7 Many examples of self-sorting can be found in the assembly of metallo-supramolecular architectures, where ligand structure,8 metal coordination,9–11 or stereochemistry12,13 can impart the resolution of complex mixtures of different metal ions and ligands into a reduced number of more complex structures. Supramolecular assemblies based on complementary ionic forces,14 H-bonding interactions15–18 or reversible covalent bond formation7,19 can also display self-sorting behaviour. Amongst bio-inspired soft-matter materials, orthogonal self-assembly of surfactants may lead to interpenetrating networks of fibrillar networks20–25 or to their co-existence with liposomes26 or liquid crystal bolaamphiphiles, whereas the combination of H-bonding and hydrophobic units can provide self-sorting in micelles.27The concept of self-sorting is crucial for understanding the formation of complex biological systems and carries great potential for bio-organic materials. However, its application to molecular electronics is still rare despite the promise of constructing complex, multi-functional devices from the designed self-assembly of smaller and simpler components.28–33 Donor–acceptor stacks exhibiting selective charge transfer properties have been obtained by co-crystallization34 or self-assembly in solution,35,36 or from organization on surfaces.37,38 In the case of perylenediimide- and stilbene-based fibrillar networks, self-sorting was shown by small-angle neutron scattering to be sensitive to gelation conditions which also affects inter-component electron transfer.39 Unfortunately, the same forces that induce self-sorting at the molecular level are generally of limited compatibility with the preparation of solution- or vacuum-processed electronic devices. For example, the use of orthogonal molecular recognition motifs may lead to the selective solubilisation or precipitation of one of the components during deposition of the active layer, whereas selection based on the use of long aliphatic chains is not generally conducive towards charge transport. While the interest in exploiting narcissistic molecular self-sorting for photovoltaic applications is obvious, a particularly attractive application of self-sorting would be in the fabrication of OLED devices incorporating pixels of different colours from a single solution of materials.
Our previous work on the use of supramolecular interactions for self-assembled OLED materials led us to the design of electroactive molecules incorporating biuret units.40 The latter present two orthogonal H-bonding sites and are known to preferentially form sheet-like structures in the solid state. We found the presence of two biuret units at the extremities of a rigid aromatic core to be a general principle guiding the formation of vesicle-like aggregates in organic solvents. Interestingly, the latter are formed spontaneously and can be relatively monodisperse in size (200–500 nm diameter).41 Upon deposition, evaporation of the solvent leads to the formation of flat disks whose dimensions are determined by the diameter and wall thickness of the aggregate in solution. Thus, disks with diameters of 200–500 nm and only 30–60 nm thick are easily obtained. TEM analysis of the edges of drop-cast samples evidences the formation of multi-walled tubes that undergo constriction to give hollow spheres whose size is directed by the diameter of the tube. Such tubes, formed by rolling up of the H-bonded sheets, would place the H-bonding motifs in the plane of the sphere's surface in agreement with static light scattering results indicating the absence of strong aggregate–aggregate interactions in solution.41
Results and discussion
The formation of vesicle-like aggregates from biuret-appended π-conjugated cores is robust, and is a typical case of social self-sorting. Thus, mixing a varying amount of a compound with lower excitation energy gives rise to efficient inter-chromophore energy transfer resulting from fast exciton migration and high density of chromophores. In this fashion, it is possible to obtain aggregates of different colours, including white, by mixing biuret-appended compounds with different emission profiles in solution. The resulting drop- or spin-cast materials possess narrow colour distributions, implying that the dispersion of the components is homogeneous between aggregates. Simultaneously obtaining aggregates of different colours in a single device is therefore not directly possible using this approach and requires either the sequential deposition of the different colours42,43 or the use of a colour-bleaching technique44 to subsequently tune the emission profile of the material. Although the size of the rigid, π-conjugated core does not seem to have a strong effect on the formation of the vesicle-like aggregates, we remarked that this is not the case for the orientation of the biuret molecular recognition motifs. Indeed, reversible conversion between aggregates exhibiting different morphologies (vesicles and fibers) could be obtained through photoisomerization of an azobenezene-containing core. This result prompted us to probe the influence of the connecting point (meta vs. para) between the biuret unit and the conjugated chromophore, revealing that some of the components of these two systems can undergo narcissitic self-sorting, which could be used to obtain solid-state OLED devices with pixels of different colors from a single solution of chromophores. We rationalize this behaviour in terms of the packing of molecules within the aggregate shell.The synthesis of compounds 1–3 in their para- and meta- series through Suzuki cross-coupling of 3- or 4-biuretphenylboronic acid with the corresponding aromatic core was described previously.40,44 Both series of compounds spontaneously generate vesicle-like aggregates upon dissolution in anhydrous THF (10−4 M concentration) that, upon deposition on conductive substrates, allows the systematic fabrication of OLED devices emitting in the blue, green, and red regions of the visible spectrum.42,44 Dynamic and static light scattering experiments, and small angle X-ray diffraction are consistent with the presence, in solution, of hollow sphere aggregates possessing a thin (ca. 30 nm) shell that are subject to Brownian motion (i.e. that do not interact with one another).41
Mixing compounds p1–p3 or m1–m3 in solution results in the deposition of disk-like aggregates that can emit a single colour due to efficient energy migration and transfer processes along with a homogeneous dispersion of the compounds in the aggregates. This is shown in Fig. 1, where the reconstructed colour confocal fluorescence microscope images of the aggregates of pure m1, m2, and m3 (calculated from the corrected emission spectra) are shown. From the localization of the emission profiles on the chromatic CIE chart (Fig. 1F), it can be seen that the emissions are very monodisperse and localized in the blue, green, or red areas of the visible region. The emission of m1, but containing 1% of m3 also gives a homogeneous dispersion of aggregates whose emission is localized between the CIE colour coordinates of m1 and m3 (Fig. 1D). In the case of m2 doped with m3 (10%, Fig. 1E), the emission is now shifted towards the yellow with some small differences between individual aggregates. This is indicative of some narcissistic self-sorting even between such structurally similar compounds (see also Fig. S7 in the ESI‡). The changes in the emission profile in the samples shown in Fig. 1D and E compared to those in Fig. 1A and B are attributed to efficient energy transfer to m3 from excited m1 or m2, coupled to fast exciton migration in the aggregates. This allows the donor excited state to sample numerous sites, any one of which may undergo radiative energy transfer to a nearby m3 molecule. Such processes are known to be highly efficient and quenching efficiencies in excess of one acceptor for 100 donors have been observed in fibrillar aggregates.45
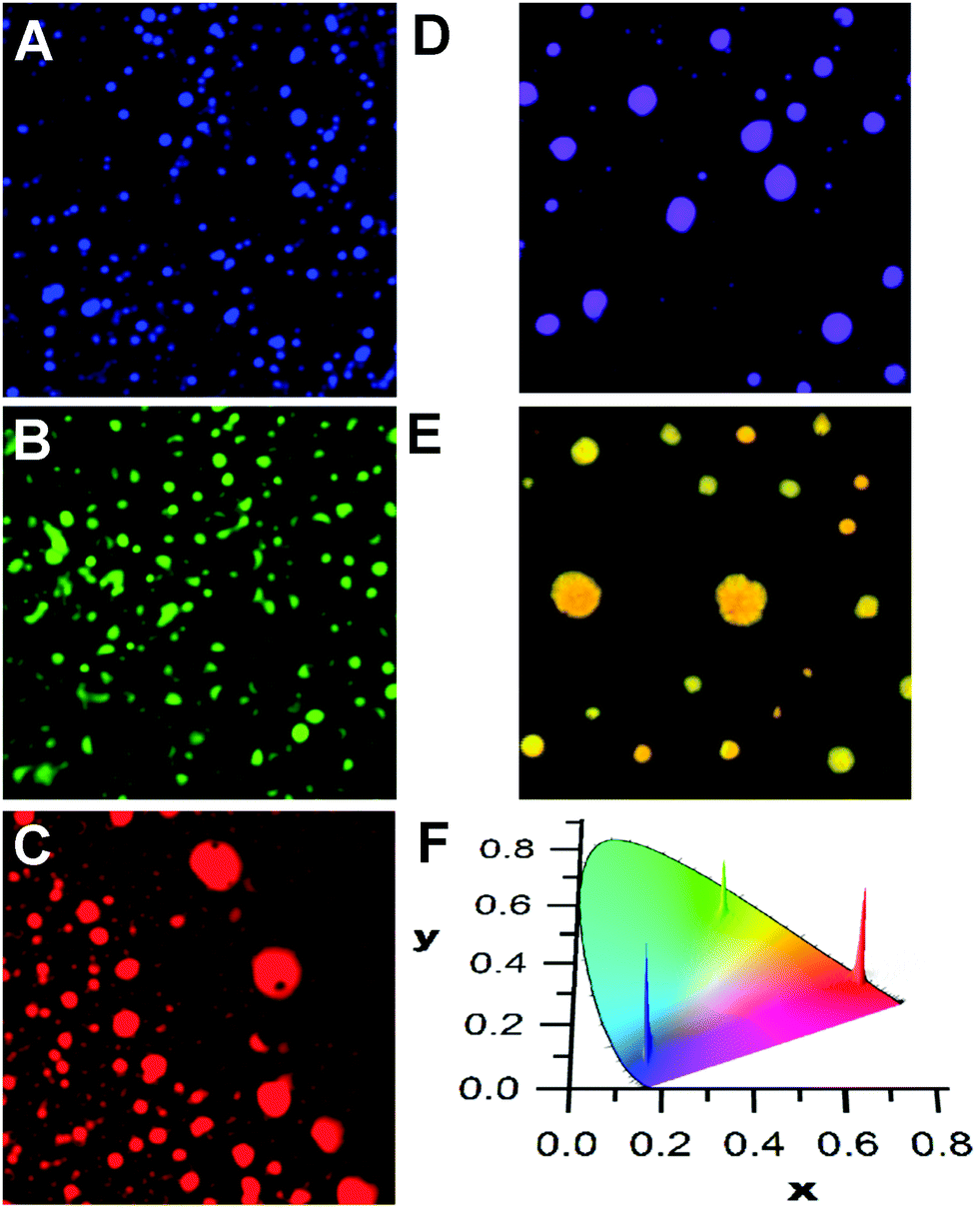 | ||
Fig. 1 Reconstructed colour images of aggregates deposited from solutions (10−4 M in THF) of m1 (A), m2 (B), and m3 (C) and of mixtures of m1 and m3 (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10−4 M in THF, panel (D)) or of mixtures of m2 and m3 (100:10, 10−4 M in THF, panel (E)). The dispersions of colours from the emission of the aggregates from A–C are shown in the CIE chart (F). Images are 20 × 20 μm (λex = 375 nm). :1, 10−4 M in THF, panel (D)) or of mixtures of m2 and m3 (100:10, 10−4 M in THF, panel (E)). The dispersions of colours from the emission of the aggregates from A–C are shown in the CIE chart (F). Images are 20 × 20 μm (λex = 375 nm). | ||
A similar social self-sorting behaviour is found for most, but not all, combinations of compounds shown in Scheme 1. It is interesting to note that the combination of m2 and m3 gives aggregates that can display a small degree of narcissistic self-sorting despite the similarities of the structures of m2 and m3. The propensity towards narcissistic self-sorting is increased when m3 is replaced by p3 (Fig. 2). Thus, blending of m1 (blue) or m2 (green) with p3 (red) gives rise to a mixture of aggregates with a clear two-tone color distribution. In the case of a solution of m1 and p3 (100:1, 10−4 M in THF), a dispersion of blue and magenta aggregates can be observed (Fig. 2A).
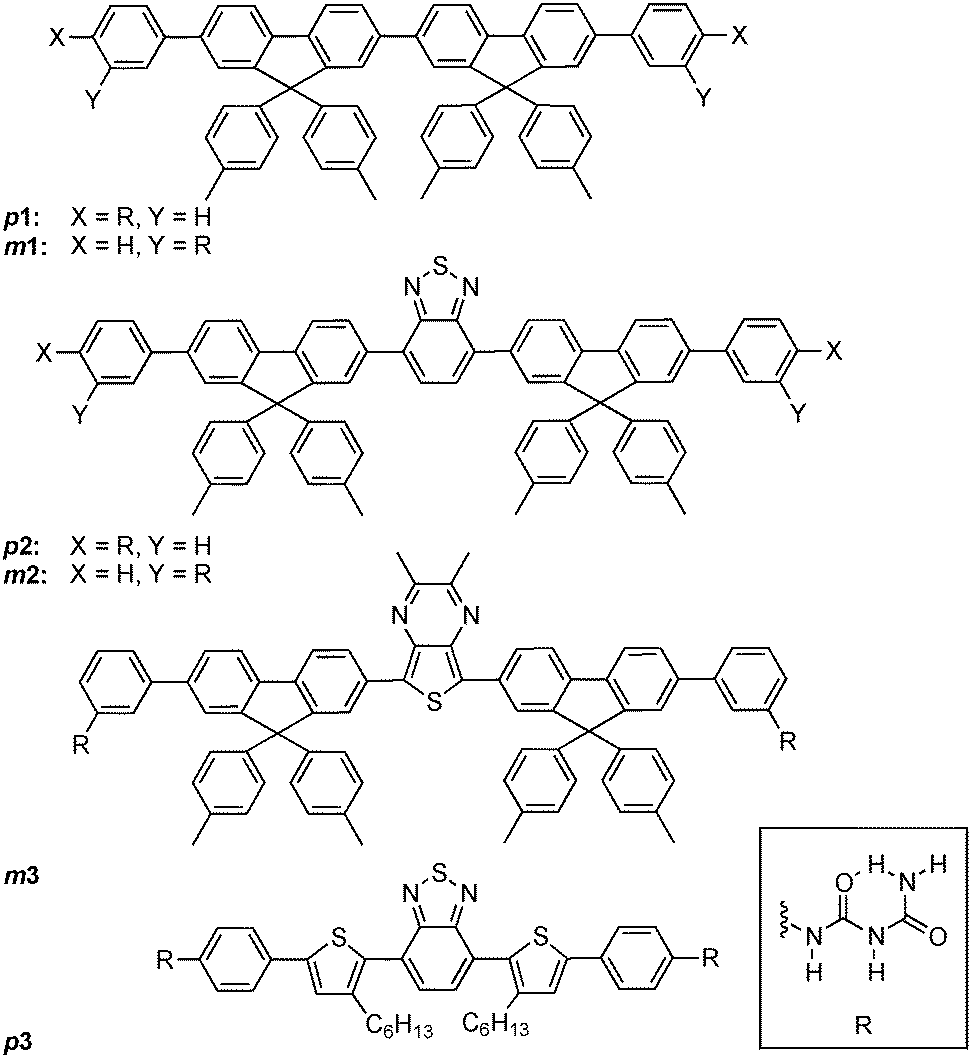 | ||
| Scheme 1 Structures of compounds used in self-sorting experiments. | ||
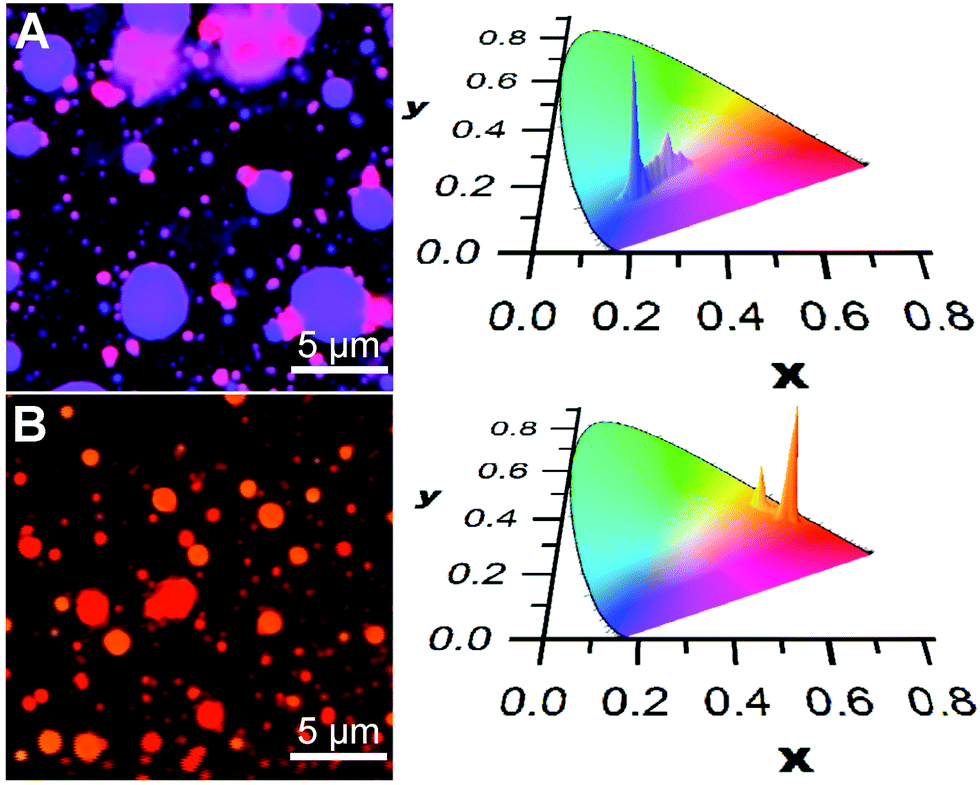 | ||
| Fig. 2 Reconstructed colour image (A) of aggregates deposited from solutions of m1 and p3 (100:1, 10−4 M in THF), and reconstructed colour image (B) of aggregates deposited from solutions of m2 and p3 (100:10, 10−4 M in THF). On the right are shown the CIE plots of the distributions of colours. The inhomogeneous dispersions of p3 into m1 or m2 are examples of narcissistic self-sorting (λex = 375 nm). | ||
Blending of m2 with p3 also results in narcissistic self-sorting, as evidenced by the results shown in Fig. 2B. In this case, deposition of a solution containing both m2 and p3 (10:1, 10−4 M in THF) gives rise to a distribution of vesicle-like aggregates that emit in the orange or red region. Although the difference between the two appears small to the naked eye due to its low sensitivity in this region of the spectrum, it is much more apparent in the CIE chromatic diagram showing the distributions of colour pixels. Here, it is clear that the distribution between the two components is much better defined than in the m1/p3 system, giving rise to two distinct emission distribution centred around the emission of each of the two aggregates' emission profiles. However, in contrast to the m1/p3 system, fluorescence lifetime imaging does not reveal significant differences in the emission decay parameters between the aggregates as these possess a broad distribution of lifetimes centered at ca. 5 ns.§
To evaluate the degree of narcissistic self-sorting, we compared the emission spectra of the self-sorted components with those of model systems displaying social self-sorting. In the case of the m1/p3 system, we selected the m1/m3 combination as a reference. The absorption and emission profiles of p3 and m3 are similar, and we can therefore expect that the efficiency of energy transfer should be comparable. Solutions containing m1 and m3 give rise to aggregates for which the emission is homogeneous throughout the sample. Provided that this implies that the concentrations of the components are also homogeneous, this allows us to estimate the relative proportion of each component in the self-sorted vesicles (see the ESI‡ for details). In the case of the m1/p3 system, we estimate the ratio of the blue to the red component to be 100:0.4 and 100:9 for the blue and magenta aggregates, respectively. This gives a self-sorting ratio of 9/0.4 = 23. In the case of the m2/p3 system, a similar analysis gives a composition of green to red component of 100:4 and ca. 100:19 for the orange and red aggregates, respectively.¶ It should be noted, however, that despite the lower self-sorting ratio for the m2/p3vs.m1/p3 system, the former has a clearer separation between the colour components resulting from a less broad distribution compared to the m1/p3 system for which the distribution in colours of the magenta component is significantly broader.
Analysis of the decay kinetics of the donor and acceptor components in the self-sorted vesicles provides additional information concerning how the molecules are organized within individual vesicles. Although single vesicles are close in size to the diffraction-limited resolution of the microscope (ca. 200 nm), the distribution of decay constants that is collected reflects the different environments of the emitting species. Thus, in the case of the blue-emitting vesicles in the self-sorted systems of m1 and p3 (100:1, 10−4 M in THF), we find that the decay of m1 exhibits a dual distribution of lifetimes indicating that the environment of m1 inside the vesicle is not uniform (Fig. 3). One of the distributions, centered at ca. 1.3 ns, is similar to that of the unquenched species, which we attribute to the presence of patches of pure or nearly-pure m1 in the vesicle. The shorter component is instead attributed to regions of the vesicles in which some p3 is present, resulting in quenching of the emission. Not unexpectedly, imaging the lifetime distribution on the vesicle surface area does not readily reveal regions in which different domains are visible as these would be below the instrumental resolution. Analysis of the acceptor decay kinetics shows the expected symmetrical distribution of lifetimes centred at the mean lifetime of the unquenched acceptor (4–5 ns). In the case of the m2/p3 system, unsymmetrical decay distributions are also seen, but the overlap in emission between the donor and acceptor does not allow us to acquire and analyse each decay component individually.
 | ||
| Fig. 3 Fluorescence lifetime imaging (λex = 375 nm) of a single blue-emitting aggregate deposited from solutions of m1 and p3 (100:1, 10−4 M in THF) collected at 400–450 nm (A) or 570–650 nm (B). In (A), only the emission from m1 is observed and it exhibits a dual distribution of lifetimes in agreement with the existence of regions (blue patches) where no quenching by p3 is observed. The emission from p3 (B) shows a single lifetime distribution as expected for direct or sensitized excitation. | ||
The origin of the self-sorting behaviour of only a few of the 12 possible combinations is intriguing. Clearly, the location of the biuret motif (meta vs. para) is important, since varying only the core (e.g.p1/p3 or p2/p3) does not lead to significant self-sorting despite the identical difluorene-based chromophores used for 1 and 2. Likewise, only varying the point of attachment is not sufficient since not all of the para/meta combinations give rise to self-sorting. Our previous investigations of the self-assembly process in solution are consistent with a model in which the shell of the vesicles is composed of molecules that are oriented with their long axis parallel to the surface.41 This would favour H-bonding resulting from ordered packing of the molecules. It is plausible that self-sorting results from the difference in packing arrangement of the p3 molecules with respect to the m1 and m2 compounds due to the combined difference in the position of the biuret and the structure of the core.
Conclusions
The spontaneous aggregation behavior of a series of rigid chromophores appended with biuret H-bonding motifs provides examples of both social and narcissistic self-sorting. The former is more pronounced, as would be expected based on the structural similarity of the molecular constituents. This results in the formation of disk-like aggregates whose emission envelope was previously shown to be tunable over the visible range of the spectrum. Interestingly, even a minor structural modification resulting from exchanging a benzothiodiazole with a thienopyriazene spacer (m2vs.m3) inside the hexaphenyl aromatic core is sufficient to induce a small degree of narcissisitic self-sorting. This is amplified by changing the point of attachment of the biuret molecular recognition motif, which results in some systems displaying stronger narcissistic self-sorting behaviour. Such sensitivity to the molecular structure is likely the result of structural order in the vesicle shell that is itself highly dependent on molecular structure. Prediction of which structural modification will lead to a specific sorting behaviour remains, however, elusive at present. Even so, this behaviour can give rise to the spontaneous formation of side-by-side aggregates emitting different colors from deposition of a single solution of both components. This approach could provide a solution to mitigate the effect of colour bleeding in the fabrication of OLEDs, which is a limiting factor in determining the resolution and brightness of commercial devices. In this respect, this example represents an important achievement by demonstrating the importance of even minor changes in the molecular structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from LabEx AMADEus (ANR-10-LABX-0042-AMADEUS through grant ANR-10-IDEX-0003-02) and the Agence Nationale de la Recherche - Ministry of Science and Technology (MOST) Taiwan joint funding (grants ANR-17-CE24-0033 and 107-2923-M-002-001-MY3).Notes and references
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed.
- R. Kramer, J. M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CrossRef CAS PubMed.
- M. J. Mayoral, C. Rest, J. Schellheimer, V. Stepanenko and G. Fernandez, Chemistry, 2012, 18, 15607–15611 CrossRef CAS PubMed.
- P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef CAS.
- J.-F. Ayme and J.-M. Lehn, Adv. Inorg. Chem., 2018, 71, 3–78 CrossRef CAS.
- M. Kolodziejski, A. R. Stefankiewicz and J.-M. Lehn, Chem. Sci., 2019, 10, 1836–1843 RSC.
- J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS PubMed.
- J. Zhong, L. Zhang, D. P. August, G. F. S. Whitehead and D. A. Leigh, J. Am. Chem. Soc., 2019, 141, 14249–14256 CrossRef CAS PubMed.
- W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC.
- F. De Campo, D. Lastecoueres, J.-M. Vincent and J.-B. Verlhac, J. Org. Chem., 1999, 64, 4969–4971 CrossRef CAS PubMed.
- D. M. Bassani, J.-M. Lehn, K. Fromm and D. Fenske, Angew. Chem., Int. Ed., 1998, 37, 2364–2367 CrossRef CAS.
- I. Pianet and J.-M. Vincent, Inorg. Chem., 2004, 43, 2947–2953 CrossRef CAS PubMed.
- S.-J. Rao, Q. Zhang, J. Mei, X.-H. Ye, C. Gao, Q.-C. Wang, D.-H. Qu and H. Tian, Chem. Sci., 2017, 8, 6777–6783 RSC.
- D. Ajami, J.-L. Hou, T. J. Dale, E. Barrett and J. Rebek, Jr., Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10430–10434 CrossRef CAS PubMed.
- D. S. Philips, K. K. Kartha, A. T. Politi, T. Krueger, R. Q. Albuquerque and G. Fernandez, Angew. Chem., Int. Ed., 2019, 58, 4732–4736 CrossRef CAS PubMed.
- A. Nuthanakanti, M. B. Walunj, A. Torris, M. V. Badiger and S. G. Srivatsan, Nanoscale, 2019, 11, 11956–11966 RSC.
- S. Schoder and C. A. Schalley, Chem. Commun., 2017, 53, 9546–9549 RSC.
- K. Acharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed.
- Y. Wang, M. Lovrak, Q. Liu, C. Maity, V. A. A. le Sage, X. Guo, R. Eelkema and J. H. van Esch, J. Am. Chem. Soc., 2019, 141, 2847–2851 CrossRef CAS PubMed.
- Y. Wang, R. M. de Kruijff, M. Lovrak, X. Guo, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2019, 58, 3800–3803 CrossRef CAS PubMed.
- W. Tanaka, H. Shigemitsu, T. Fujisaku, R. Kubota, S. Minami, K. Urayama and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 4997–5004 CrossRef CAS PubMed.
- B. O. Okesola, Y. Wu, B. Derkus, S. Gani, D. Wu, D. Knani, D. K. Smith, D. J. Adams and A. Mata, Chem. Mater., 2019, 31, 7883–7897 CrossRef CAS PubMed.
- G. Liu, C. Zhou, W. L. Teo, C. Qian and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 9366–9372 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Nat. Chem., 2016, 8, 737–738 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, M. C. A. Stuart, L. Florusse, G. Raffy, A. Del Guerzo and J. H. van Esch, Chem. Sci., 2016, 7, 6021–6031 RSC.
- A. Pal, S. Karthikeyan and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 7842–7843 CrossRef CAS PubMed.
- H. Kar and S. Ghosh, Isr. J. Chem., 2019, 59, 881–891 CrossRef CAS.
- K.-T. Wong and D. M. Bassani, NPG Asia Mater., 2014, 6, e116 CrossRef CAS.
- E. R. Draper, B. Dietrich and D. J. Adams, Chem. Commun., 2017, 53, 1864–1867 RSC.
- E. R. Draper, J. R. Lee, M. Wallace, F. Jackel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC.
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS PubMed.
- W.-C. Geng, Y.-C. Liu, Y.-Y. Wang, Z. Xu, Z. Zheng, C.-B. Yang and D.-S. Guo, Chem. Commun., 2016, 53, 392–395 RSC.
- M. A. Niyas, R. Ramakrishnan, V. Vijay and M. Hariharan, Chem. – Eur. J., 2018, 24, 12318–12329 CrossRef CAS PubMed.
- V. S. Nair, R. D. Mukhopadhyay, A. Ajayaghosh, A. Saeki and S. Seki, Sci. Adv., 2016, 2, e1600142 CrossRef PubMed.
- C.-H. Huang, N. D. McClenaghan, A. Kuhn, J. W. Hofstraat and D. M. Bassani, Org. Lett., 2005, 7, 3409–3412 CrossRef CAS PubMed.
- N.-T. Lin, A. Vargas Jentzsch, L. Guenee, J.-M. Neudoerfl, S. Aziz, A. Berkessel, E. Orentas, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1121–1127 RSC.
- A. Mendez-Ardoy, N. Markandeya, X. Li, Y.-T. Tsai, G. Pecastaings, T. Buffeteau, V. Maurizot, L. Muccioli, F. Castet, I. Huc and D. M. Bassani, Chem. Sci., 2017, 8, 7251–7257 RSC.
- E. R. Cross, S. Sproules, R. Schweins, E. R. Draper and D. J. Adams, J. Am. Chem. Soc., 2018, 140, 8667–8670 CrossRef CAS PubMed.
- K.-P. Tseng, F.-C. Fang, J.-J. Shyue, K.-T. Wong, G. Raffy, G. A. Del and D. M. Bassani, Angew. Chem., Int. Ed., 2011, 50, 7032–7036 CrossRef CAS PubMed.
- S. K. P. Velu, M. Yan, K.-P. Tseng, K.-T. Wong, D. M. Bassani and P. Terech, Macromolecules, 2013, 46, 1591–1598 CrossRef CAS.
- Y.-T. Tsai, K.-P. Tseng, Y.-F. Chen, C.-C. Wu, G.-L. Fan, K.-T. Wong, G. Wantz, L. Hirsch, G. Raffy, A. Del Guerzo and D. M. Bassani, ACS Nano, 2016, 10, 998–1006 CrossRef CAS PubMed.
- K.-P. Tseng, Y.-T. Tsai, J.-J. Shyue, G. Raffy, A. Del Guerzo, K.-T. Wong and D. M. Bassani, Chem. Phys. Lett., 2017, 683, 43–48 CrossRef CAS.
- Y.-T. Tsai, H.-F. Liu, B.-J. Peng, K.-P. Tseng, M.-C. Kuo, K.-T. Wong, G. Wantz, L. Hirsch, G. Raffy, A. Del Guerzo and D. M. Bassani, ACS Appl. Mater. Interfaces, 2017, 9, 36045–36052 CrossRef CAS PubMed.
- C. Giansante, G. Raffy, C. Schäfer, H. Rahma, M.-T. Kao, A. G. L. Olive and A. D. Guerzo, J. Am. Chem. Soc., 2011, 133, 316–325 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. J.-M. Lehn on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Details of experimental setup for confocal microscopy and fluorescence lifetime images of pristine systems. See DOI: 10.1039/c9qm00636b |
| § The average lifetimes of m1, m2 and m3 in the aggregates are 1.6, 3.4 and 2.5 ns, respectively (λex = 375 nm). |
| ¶ Due to the occurrence of narcissistic self-sorting at higher loadings of m3, the proportion of m2 to p3 in the red vesicles was estimated from extrapolation of the data from m2 and m3 at lower loading ratios. |
| This journal is © the Partner Organisations 2020 |