
Design strategies of n-type conjugated polymers for organic thin-film transistors
Ying
Sui
a,
Yunfeng
Deng
*a,
Tian
Du
a,
Yibo
Shi
a and
Yanhou
Geng
 *ab
*ab
aSchool of Materials Science and Engineering, Tianjin University, Tianjin 300072, P. R. China. E-mail: yunfeng.deng@tju.edu.cn; yanhou.geng@tju.edu.cn
bTianjin Key Laboratory of Molecular Optoelectronic Science, Tianjin University, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, P. R. China
First published on 12th July 2019
Abstract
Significant progress in n-type conjugated polymers (CPs) for organic thin-film transistors (OTFTs), which are required in making complementary metal oxide semiconductor (CMOS)-like logic circuits together with p-type CPs, has been made in the last two decades by developing novel building blocks and optimizing device structures as well as processing conditions. However, the device performance, in terms of mobility and air stability, of n-type CPs is still much lower than that of their p-type counterparts. CPs play a key role in determining the device performance. The properties of CPs, such as molecular packing structures and frontier molecular orbital energy levels, can be appropriately adjusted by molecular design. In the current review, we summarize the progress of n-type CPs from the aspect of molecular design, and four molecular design strategies, i.e., donor–acceptor (D–A) CPs with strong A units, D–A CPs with weak D units, D–A CPs with dual-A and A–A type CPs are discussed. It has been demonstrated that D–A CPs with weak D units and A–A type CPs are highly desirable in the construction of unipolar n-type CPs because they generally have low-lying highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels.
 Ying Sui | Ying Sui received her BS degree in Materials Science and Engineering from Tianjin University in 2015. Now she is a PhD candidate at the School of Materials Science and Engineering, Tianjin University. She mainly researches on the synthesis of conjugated polymers by direct arylation polycondensatioin. |
 Yunfeng Deng | Yunfeng Deng received his PhD degree in Polymer Chemistry and Physics from Changchun Institute of Applied Chemistry, Chinese Academy Of Sciences in 2015. He then joined the University of Waterloo and Technion-Israel Institute of Technology as a postdoctoral research fellow. Currently, he is an associate professor in the School of Materials Science and Engineering, Tianjin University, China. His research mainly focuses on the design and synthesis of conjugated polymers and their related electronic devices. |
 Tian Du | Tian Du received his BS degree in Material Chemistry from Zhengzhou University in 2017. He then moved to Tianjin University to pursue his PhD degree. His research interests include molecular design and synthesis of π-conjugated materials and their applications in thin-film transistors. |
 Yibo Shi | Yibo Shi received his BS degree in Materials Science and Engineering from Tianjin University in 2018. He is currently pursuing his PhD degree at the School of Materials Science and Engineering, Tianjin University. His research interests mainly focus on organic semiconducting polymers. |
 Yanhou Geng | Yanhou Geng received his BS degree from Shanghai Jiaotong University in 1991 and PhD degree from Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences (CAS) in 1996. From 1998 to 2003, he worked in the Max-Planck Institute for Polymer Research in Mainz (Germany) as an AvH stipendiat and the University of Rochester (USA) as a postdoctoral researcher. From 2003 to 2015, he was a professor in the State Key Laboratory of Polymer Physics and Chemistry, CIAC. Currently, he is a full professor at the School of Materials Science and Engineering, Tianjin University, China. He has published over 200 papers in peer-reviewed journals and more than 20 patents. His research interests include (1) polymerization methods for conjugated polymers; (2) organic and polymeric semiconductors for solar cells and thin-film transistors. |
Introduction
Organic thin-film transistors (OTFTs) based on conjugated polymers (CPs) have attracted considerable attention due to their applications in low-cost, large-area and flexible electronics, such as flexible displays and wearable devices.1–12 Polymer semiconductors are the key component of OTFTs and the recent advancement of CPs promotes the application of these devices.6–9,12 According to the polarity of the dominant charge carrier in the transistor channel, CPs can be classified as p-type (hole dominant), n-type (electron dominant) and ambipolar (both carriers) semiconductors. In the past several years, a significant amount of high-performance p-type and ambipolar CPs have been reported. In contrast, there has been a paucity of n-type CPs.13–21 High-performance n-type CPs are crucial to fabricate low-power complementary metal oxide semiconductor (CMOS)-like logic circuits, which is the most important and widely used configuration in integrated circuits.21–24 Therefore, the scarcity of n-type CPs is one of the most critical issues to be addressed to realize the application of OTFTs in electronic devices.For n-type CPs, both low-lying highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels are necessary. Empirically, a HOMO energy level of <−5.9 eV and a LUMO energy level of ∼−4.0 eV are required for a stable n-type organic semiconductor.14,25–30 The low-lying HOMO energy level could build a larger hole injection barrier to block hole injection from the commonly used Au electrodes, realizing unipolar n-type transport. Meanwhile, the low-lying LUMO energy level would facilitate electron injection. Although low work function electrodes, such as Al, Ca and Mg, have been proved to help electron injection, these metals are not environmentally stable.17,21 More importantly, the low-lying LUMO energy level is beneficial for stabilizing electron transport under air conditions.18,21,25,26 It has been revealed that the air instability of n-type OTFT devices is not due to the degradation of materials, but arises from the charge carrier trapping by water and oxygen.17,25 The deep LUMO energy level could prevent the electron from being captured by water or oxygen in air and improve the air stability of the n-type OTFT device. However, the LUMO energy level cannot be far lower than −4.0 eV, because the polymer will be unintentionally “air-doped” if the LUMO energy level is too low (≤−4.5 eV), leading to the loss of its semiconductor properties.20 Therefore, for n-type CPs, a LUMO energy level between −4.0 and −4.4 eV would be desirable for air-stable electron transport. Additionally, HOMO and LUMO distribution can also influence the polarity of charge carriers of CPs. Generally, polymers with delocalized LUMOs and segregated HOMOs tend to display n-type transport characteristics. The main transport pathway of the charge carriers in an organic semiconductor is hopping through the frontier molecular orbitals. Well distributed LUMOs will facilitate LUMO–LUMO interactions, thus electron can easily move from one polymer chain to another, while localized HOMOs may result in less efficient HOMO–HOMO interactions, blocking hole hopping.31–34
Donor–acceptor (D–A) polymers, in which the D unit and A unit are alternately linked along the polymer backbone, are promising candidates for high mobility polymers due to their strong intermolecular interactions and small π–π stacking distance enabled by the large dipole moment between the D and A units. However, the electron-rich D unit, such as thiophene and bithiophene, would raise the HOMO energy levels of the resultant D–A polymers through orbital hybridization between the D and A units.35–37 The elevated HOMO energy level would reduce the hole injection barriers. As a result, the majority of D–A polymers tends to exhibit ambipolar or merely p-type charge transport characteristics (depending on the LUMO energy level). Moreover, for D–A polymers, the HOMOs are usually well delocalized along the polymer chains while the LUMOs are mostly localized on the A units,38–43 which is detrimental to both intrachain and interchain electron transport. Therefore, to achieve high-performance n-type CPs, reasonable design of D–A polymers is required or new polymer backbone design strategies should be developed.
Besides the intrinsic properties of the semiconductor polymers, the OTFT device architecture also exerts an influence on the observed device performance. There are three commonly-used OTFT device architectures, namely, bottom-gate bottom-contact (BGBC), bottom-gate top-contact (BGTC) and top-gate bottom-contact (TGBC). The fabrication of bottom-gate devices is relatively easy since the substrate Si/SiO2 wafer also serves as a gate electrode and a dielectric layer.50 However, TGBC devices frequently exhibit higher electron mobility and better air stability than those with the bottom-gate structure due to the better contact of the semiconductor layer with the organic dielectric and the encapsulation of the dielectric layer, preventing the capture of electron by moisture and oxygen.21,50
Since the first demonstration of a high-performance n-type OTFT device based on CP in the early 2000s by using a ladder-type polymer, poly[(7-oxo-7,10H-benz[de]imidazo[4′,5′:5,6]benzimidazo[2,1-a]isoquinoline-3,4:10,11-tetrayl)-10-carbonyl] (BBL), as the semiconductor layer,44 remarkable progress in the development of n-type CPs has been made via the design and synthesis of novel polymers with appropriate HOMO and LUMO energy levels. Recently, a D–A polymer with unipolar electron mobility (μe) of up to 7.16 cm2 V−1 s−1 has been reported,45 noticeably shortening the performance gap between p- and n-type CPs. In this article, we review the progress of n-type CPs with an emphasis on the polymer design strategies. Four molecular design strategies of n-type CPs, i.e., D–A CPs with strong A units, D–A CPs with weak D units, D–A CPs with dual-A and A–A type CPs, are introduced. The relative merits of these strategies and the future development trend of n-type CPs for OTFTs are also discussed.
Design strategies of n-type CPs
1. D–A CPs with strong A units
The development of n-type CPs is somewhat restrained by the lack of strong A units that can endow D–A CPs with low-lying HOMO and LUMO energy levels. Currently, the useful A units for n-type CPs are mainly limited to naphthalene diimide (NDI), perylene diimide (PDI), bis(oxoindolinylidene)benzodifurandione and quinoidal compounds.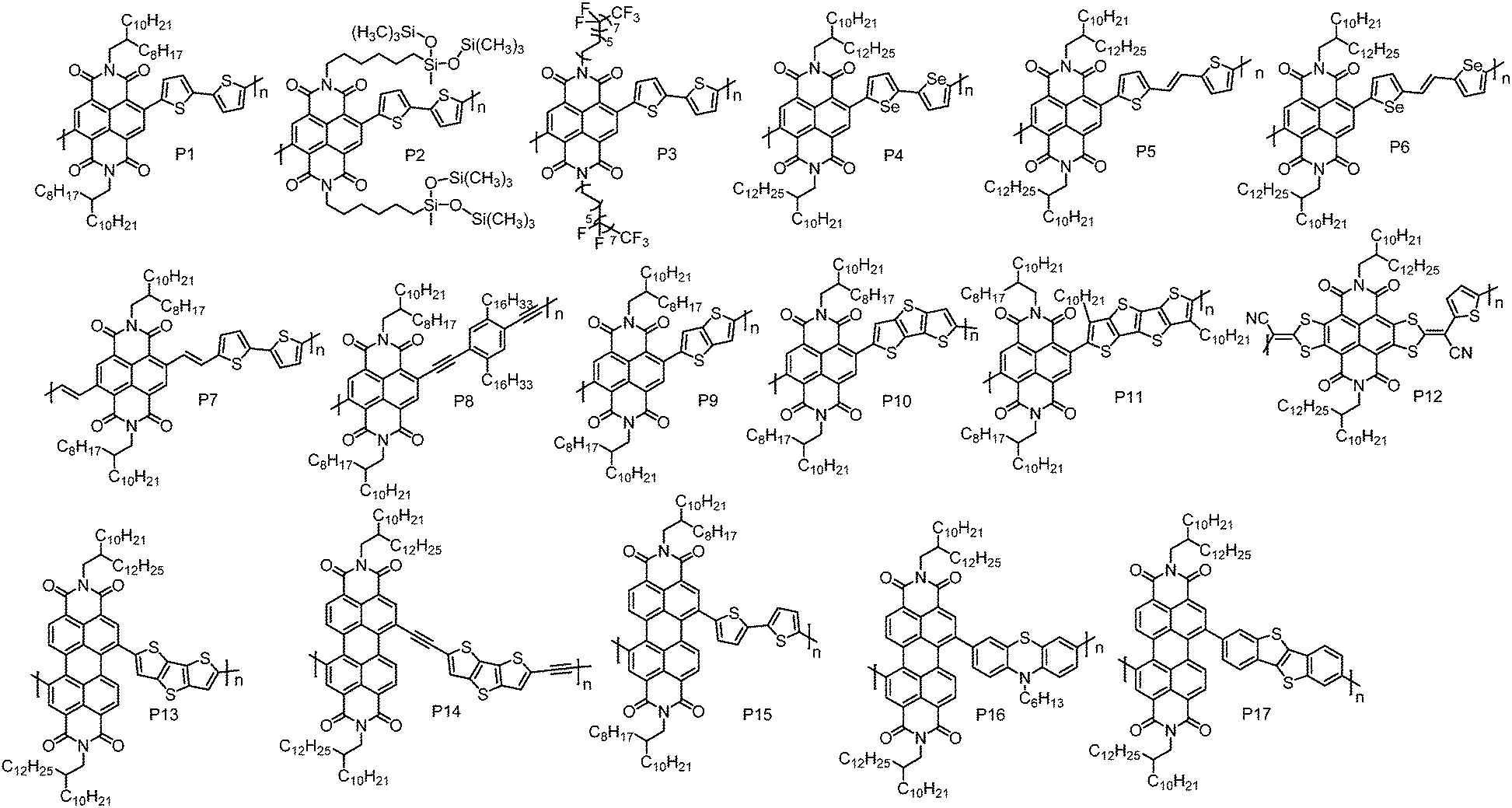 | ||
| Fig. 1 Molecular structures of the n-type D–A conjugated polymers based on NDI and PDI. | ||
| Polymers | M n (kDa) | Đ | E HOMO (eV) | E LUMO (eV) | OTFT structure | μ e (cm2 V−1 s−1) | Measured environment | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Polydispersity. b HOMO energy level measured by cyclic voltammetry, unless explicitly stated. c LUMO energy level measured by cyclic voltammetry, unless explicitly stated. d Estimated from EHOMO = ELUMO − Eoptg, in which Eoptg is the optical bandgap. e Estimated from ELUMO = EHOMO + Eoptg. f Measured by photoelectron spectroscopy. g The electrodes were modified with Cs2CO3. h The electrodes were modified with Ba(OH)2. | ||||||||
| P1 | 47.8 | 5.53 | −5.36d | −3.91 | BGTC | 0.06 | Vacuum | 47 |
| 52.5 | 5.51 | n/a | n/a | TGBC | 0.85 | Air | 48 | |
| n/a | n/a | n/a | n/a | TGBC | 1.03 | Nitrogen | 49 | |
| P2 | 32.0 | 2.02 | −5.27d | −3.83 | TGBC | 1.04 | n/a | 51 |
| P3 | 28.0 | 2.03 | −5.62d | −4.01 | BGTC | 6.50 | Nitrogen | 52 |
| P4 | 42.4 | 2.60 | −5.95 | −3.94 | BGTC | 0.07 | Nitrogen | 54 |
| P5 | 70.0 | 1.98 | −5.42d | −4.00 | TGBC | 1.80g | Nitrogen | 57 |
| P6 | 80.0 | 2.10 | −5.29d | −3.98 | TGBC | 0.50g | Nitrogen | 58 |
| P7 | 117 | 2.90 | −5.55f | −4.05e | BGTC | 0.07h | Nitrogen | 63 |
| P8 | 49.0 | 2.90 | n/a | n/a | BGTC | 3.7 × 10−3 | Nitrogen | 64 |
| P9 | 62.5 | 3.30 | −5.45d | −3.88 | BGTC | 7.0 × 10−3 | Nitrogen | 69 |
| P10 | 21.3 | 2.90 | −5.31d | −3.91 | BGTC | 4.8 × 10−3 | Nitrogen | 69 |
| P11 | 92.4 | 3.60 | −5.29d | −3.92 | BGTC | 0.012 | Nitrogen | 69 |
| P12 | 33.2 | 7.00 | −5.82 | −4.25 | BGBC | 0.38 | Nitrogen | 70 |
| P13 | 15.0 | 1.50 | −5.90 | −3.90 | BGTC | 0.013 | Nitrogen | 71 |
| P14 | 15.0 | 1.10 | −5.80 | −4.00 | BGTC | 0.06 | Air | 73 |
| P15 | 11.0 | 2.90 | −5.61d | −3.96 | BGTC | 2.0 × 10−3 | Vacuum | 47 |
| P16 | 9.97 | 1.53 | −5.85 | −3.75 | BGTC | 0.05 | Nitrogen | 74 |
| P17 | 11.1 | 1.90 | −5.71d | −3.85 | BGTC | 0.042 | Nitrogen | 75 |
Yang et al. synthesized an NDI-based polymer P2 by incorporating hybrid siloxane chains.51 The pre-aggregation of P2 in solution was highly sensitive to the solvent; thus the degrees and types of the aggregates in thin-films could be tuned by the solvent, leading to solvent-dependent mobility. With chloroform as the casting solvent, P2-based TGBC OTFT devices showed the best performance with a μe of 1.04 cm2 V−1 s−1, which was much higher than that of P1 (0.32 cm2 V−1 s−1) when the devices were fabricated under the same conditions. The high μe of P2 could be attributed to its balanced face-on and edge-on molecular orientations and strong π–π interactions as evidenced by the X-ray diffraction (XRD) analysis. An NDI-based polymer with semifluoroalkyl side chains, P3, was reported by Cho et al.52 The strong self-organization of semifluoroalkyl side chains induced the polymer backbones to form superstructures composed of ‘‘backbone crystals’’ and ‘‘side-chain crystals’’. In BGTC OTFT devices, P3 showed a μe of 3.93 cm2 V−1 s−1 and the device performance was almost unchanged when exposed to air for three months. Device optimization by adding a small fraction of 1-chloronaphthalene to the casting solvent further improved the device performance, and a μe as high as 6.50 cm2 V−1 s−1 was achieved.
Compared to sulfur, selenium has a larger atomic radius, which could promote orbital overlap and increase mobility.53 Biselenophene was incorporated into an NDI-based polymer to afford P4 by Jenekhe et al.54 Relative to the bithiophene analogue, P4 showed a red-shifted absorption spectrum and a ca. 0.1 eV lower LUMO energy level. In BGTC OTFT devices, P4 displayed a μe of 0.07 cm2 V−1 s−1, which is higher than that of the bithiophene copolymer (0.04 cm2 V−1 s−1) under the same device configuration. This can be ascribed to the enhanced crystallinity of P4 induced by the Se–Se interactions.
Thiophene–vinylene–thiophene (TVT) that has a highly coplanar structure can also promote polymer interchain packing and reduce the π–π stacking distance.55,56 By incorporating a TVT unit, Kim et al. synthesized P5.57 It formed highly ordered thin-films that featured strong π–π intermolecular interactions. However, the existence of the vinylene unit slightly raised the HOMO energy level, reducing the hole injection barrier. Consequently, an electron dominant transport characteristic with weak hole transport was observed in P5-based TGBC OTFT devices. When the Au electrodes were modified with Cs2CO3, P5 exhibited unipolar n-type transport with the highest μe of up to 1.80 cm2 V−1 s−1. Selenophene–vinylene–selenophene (SVS) also has been used as the comonomer in an NDI-based polymer (P6) by the same group.58 Similarly, P6 showed an electron dominant transport characteristic with a μe of up to 2.4 cm2 V−1 s−1 in TGBC OTFT devices, which was better than that of P5. However, the Cs2CO3 treated unipolar n-type OTFT devices based on P6 exhibited a low μe of 0.50 cm2 V−1 s−1.
There is a considerable torsion angle (ca. 40°) between NDI and the adjacent thiophene units.59–62 This twist may hamper frontier molecular orbital delocalization and π–π stacking of the polymer backbones, thereby impairing electron transport. Efforts have been made to promote the backbone planarity. Heeney et al. found that insertion of a vinylene spacer between NDI and thiophene units could significantly reduce the torsion angle from 48° to 18°.63 As a result, the polymer with the vinylene spacer (P7) showed a red-shifted UV-vis absorption spectrum and a denser lamellar structure with a π–π stacking distance of 3.5 Å compared to P1. In BGTC OTFT devices with Ba(OH)2 modified Au electrodes, P7 exhibited unipolar n-type transport with the highest μe of 0.07 cm2 V−1 s−1. An NDI-based polymer with an ethynylene spacer, P8, was also synthesized by Marder et al. This polymer showed inferior performance with a μe of 3.70 × 10−3 cm2 V−1 s−1.64
A fused π system can endow molecules with greater π-orbital overlap and provide a more efficient pathway for intermolecular charge transfer.34,65–68 Luscombe et al. reported a series of NDI-based polymers with fused-thiophene units, namely, P9, P10 and P11.69 In order to keep good solution processability, linear n-dodecyl chains were installed on quaterthiophene (P11). Surprisingly, these polymers exhibited poor performance compared to the polymer with a nonfused-thiophene unit. Among these polymers, P11 displayed the best performance with the maximum μe of 0.012 cm2 V−1 s−1 in tetramethylsiloxane-bis(benzocyclobutene) derivative (BCB) modified BGTC OTFT devices. The inferior performances of these polymers are associated with their disordered thin-films. Incorporation of fused thiophene units would reduce conformational freedom of the polymer backbone, impeding self-organization of polymer backbones into ordered microstructures. A core-extended NDI-based polymer, P12, was developed by Gao et al.70 By combining the electron-withdrawing ability of NDI and cyano groups, P12 showed a deep LUMO energy level of −4.25 eV, which is lower than those of the most reported NDI-based polymers. Despite the amorphous thin-film feature, BGBC OTFT devices of P12 still exhibited a high μe of 0.38 cm2 V−1 s−1.
PDI is another rylene diimide building block with a low-lying LUMO energy level and has gained much attention in designing n-type CPs. The first PDI-based CP, P13, was synthesized by Zhan et al., which displayed unipolar n-type transport behaviour with a μe of 0.013 cm2 V−1 s−1 in BGTC OTFT devices.71 The unipolar n-type transport characteristic of P13 is attributed to its low-lying HOMO and LUMO energy levels, which are −5.90 and −3.90 eV, respectively. An improved μe of 0.06 cm2 V−1 s−1 for P13 was reported by Liu et al. using TGBC OTFT device geometry.72 Acetylene linkages were introduced into P14 by Zhan et al. to reduce the steric hindrance between PDI and dithienothiophene units.73 Compared to P13, P14 showed a red-shifted absorption, deeper LUMO energy level and more ordered microstructures in thin-films. With the combination of a low-lying LUMO energy level and a highly ordered thin-film, P14 exhibited air stable electron transport with a high μe of 0.06 cm2 V−1 s−1 in BGTC OTFT devices. In contrast, P13-based devices did not function with the same device structure in air, which only worked in nitrogen with a low μe of 0.017 cm2 V−1 s−1. Facchetti et al. reported a PDI–bithiophene copolymer (P15), showing a μe of 2.0 × 10−3 cm2 V−1 s−1 in BGTC OTFT devices.47 By using phenothiazine as the comonomer, P16, with a μe approaching 0.05 cm2 V−1 s−1, has been synthesized by Zhan et al.74 Recently, benzothieno[3,2-b]benzothiophene (BTBT) was incorporated into a PDI-based polymer, yielding P17. In BGTC OTFT devices, P17 showed a μe of 0.042 cm2 V−1 s−1.75
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 81 (Fig. 2 and Table 2).
81 (Fig. 2 and Table 2).
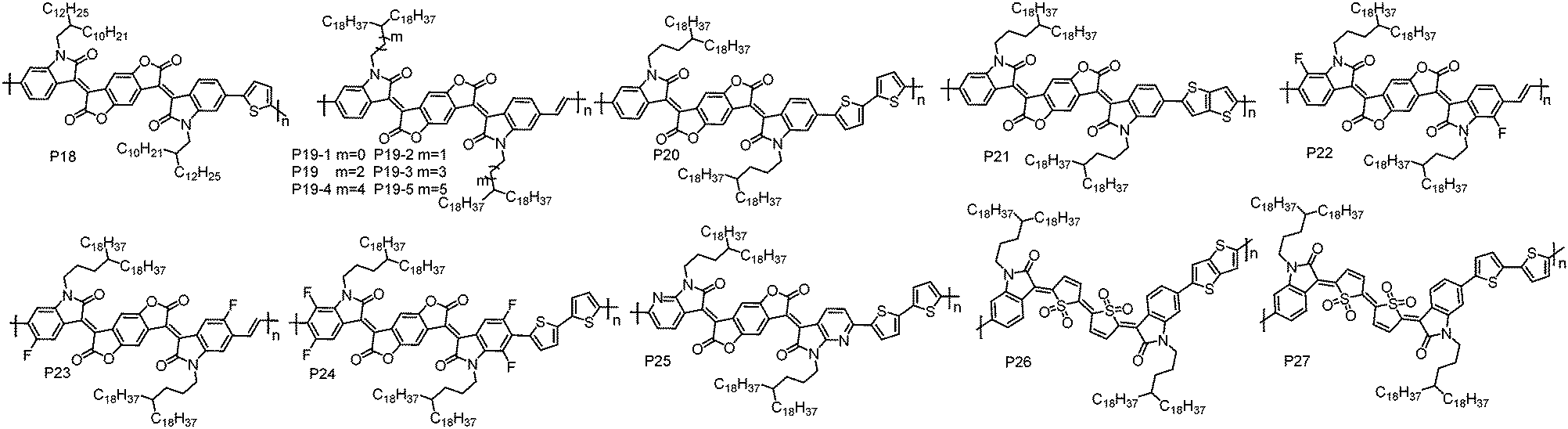 | ||
| Fig. 2 Molecular structures of the n-type D–A conjugated polymers based on BDOPV, BDOPV derivatives and an IDTO unit. | ||
| Polymers | M n (kDa) | Đ | E HOMO (eV) | E LUMO (eV) | OTFT structure | μ e (cm2 V−1 s−1) | Measured environment | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Polydispersity. b HOMO energy level measured by cyclic voltammetry, unless explicitly stated. c LUMO energy level measured by cyclic voltammetry, unless explicitly stated. d The device was fabricated with the mixture solvent of toluene and o-dichlorobenzene (1/4 vol%). e Estimated from the high VGS region. f Estimated from the low VGS region. | ||||||||
| P18 | 34.2 | 1.46 | −5.79 | −4.11 | BGBC | 5.4 × 10−3 | Air | 76 |
| P19 | 37.6 | 2.38 | −6.12 | −4.10 | TGBC | 1.10 | Air | 77 |
| 30.2 | 2.25 | −6.16 | −4.01 | TGBC | 1.40 | Air | 79 | |
| P19-1 | 37.4 | 2.04 | −6.16 | −4.01 | TGBC | 0.90 | Air | 77 |
| P19-2 | 37.7 | 2.26 | −6.15 | −4.07 | TGBC | 0.60 | Air | 77 |
| P19-3 | 34.9 | 2.01 | −6.15 | −3.97 | TGBC | 0.05 | Air | 77 |
| P19-4 | 35.9 | 2.26 | −6.16 | −4.02 | TGBC | 0.76 | Air | 77 |
| P19-5 | 35.2 | 2.44 | −6.12 | −4.01 | TGBC | 0.44 | Air | 77 |
| P20 | 77.2 | 3.00 | −5.72 | −4.15 | TGBC | 1.74; 3.20d | Air | 80 and 81 |
| P21 | 26.5 | 1.65 | −5.57 | −4.10 | BGTC | 0.65 | Vacuum | 82 |
| P22 | 66.3 | 1.94 | −6.19 | −4.26 | TGBC | 1.70 | Air | 84 |
| P23 | 53.8 | 3.06 | −6.22 | −4.30 | TGBC | 0.81 | Air | 84 |
| P24 | 38.0 | 2.74 | −5.96 | −4.32 | TGBC | 1.24;e 14.9f | Air | 85 |
| P25 | 51.6 | 2.62 | −5.80 | −4.37 | TGBC | 3.22 | Air | 86 |
| P26 | 23.5 | 2.15 | −5.92 | −3.98 | BGBC | 0.14 | Nitrogen | 93 |
| P27 | 25.9 | 2.70 | −5.78 | −4.09 | BGBC | 0.18 | Nitrogen | 95 |
Qiu et al. synthesized P21 by copolymerizing BDOPV with thienothiophene. BGTC OTFT devices based on P21 showed unipolar electron transport behaviour with the highest μe of 0.65 cm2 V−1 s−1 in high vacuum.82 However, the same polymer reported by Pei et al. showed ambipolar transport characteristics under air conditions.83 This contradiction may be explained by their different testing environment since moisture and oxygen could induce hole transport as mentioned above. These results indicate that the testing environment also influences the polarity of charge carriers.
To further lower the HOMO and LUMO energy levels, fluorine (F) atoms were introduced on the BDOPV unit. P22 and P23 with F atoms at different positions on BDOPV have been developed by Pei et al.84 Due to the strong electron-withdrawing ability of F atoms, the LUMO/HOMO energy levels of P22 and P23 are significantly lower than those of P19, which are −4.26/−6.19 eV for P22 and −4.30/−6.22 eV for P23. Additionally, compared to P19, P22 and P23 showed more ordered thin-films and smaller π–π stacking distances due to the presence of F⋯H weak intramolecular interactions. P22 exhibited a μe of 1.70 cm2 V−1 s−1 in TGBC OTFT devices under air conditions, whereas P23 showed a decreased μe of 0.81 cm2 V−1 s−1 with the same OTFT device configuration. This can be related to their different backbone conformations, leading to different interchain organization and thereby charge carrier transport ability. Afterwards, a strong electron-deficient polymer P24 was developed by Pei et al. based on four F atom substituted BDOPV.85 P24 showed LUMO/HOMO energy levels of −4.32/−5.96 eV. The TGBC OTFT devices based on P24 showed an obviously kinked |ISD|1/2–VGS plot. In the low VGS region, a high μe of 14.9 cm2 V−1 s−1 was achieved, while a much lower mobility of 1.24 cm2 V−1 s−1 in the high VGS region was obtained. The authors found that this non-linear behaviour was highly correlated with the microstructures in thin-films. Thin-films with highly ordered molecular packing tend to exhibit nonideal |ISD|1/2–VGS characteristics, whereas the less ordered thin-films usually result in linear |ISD|1/2–VGS curves in the device.
Pei et al. synthesized P25 by embedding pyridine units into BDOPV.86 The replacement of benzene rings with pyridine units not only lowers the LUMO energy level, but also improves the planarity of polymer backbones because an imine-type nitrogen atom (I–N) is less sterically demanding than a CH moiety.87 A typical n-type transport characteristic with the highest μe of 3.22 cm2 V−1 s−1 was achieved in P25-based TGBC OTFT devices. Compared with P20, the enhanced performance of P25 can be ascribed to its lower LUMO energy level and less conformational disorder.
2. D–A CPs with weak D units
As we discussed above, D–A CPs tend to exhibit p-type or ambipolar transport characteristics due to their relatively high HOMO energy levels that originate from the electron-rich D unit. In order to address this issue, a strategy of constructing D–A polymers with a weak D unit has been developed, in which the weak D unit could help maintain a low HOMO energy level. Generally, incorporating electron-withdrawing groups, such as F atoms, I–N atoms and cyano groups (CN), into the electron-rich moieties can deliver the weak D units.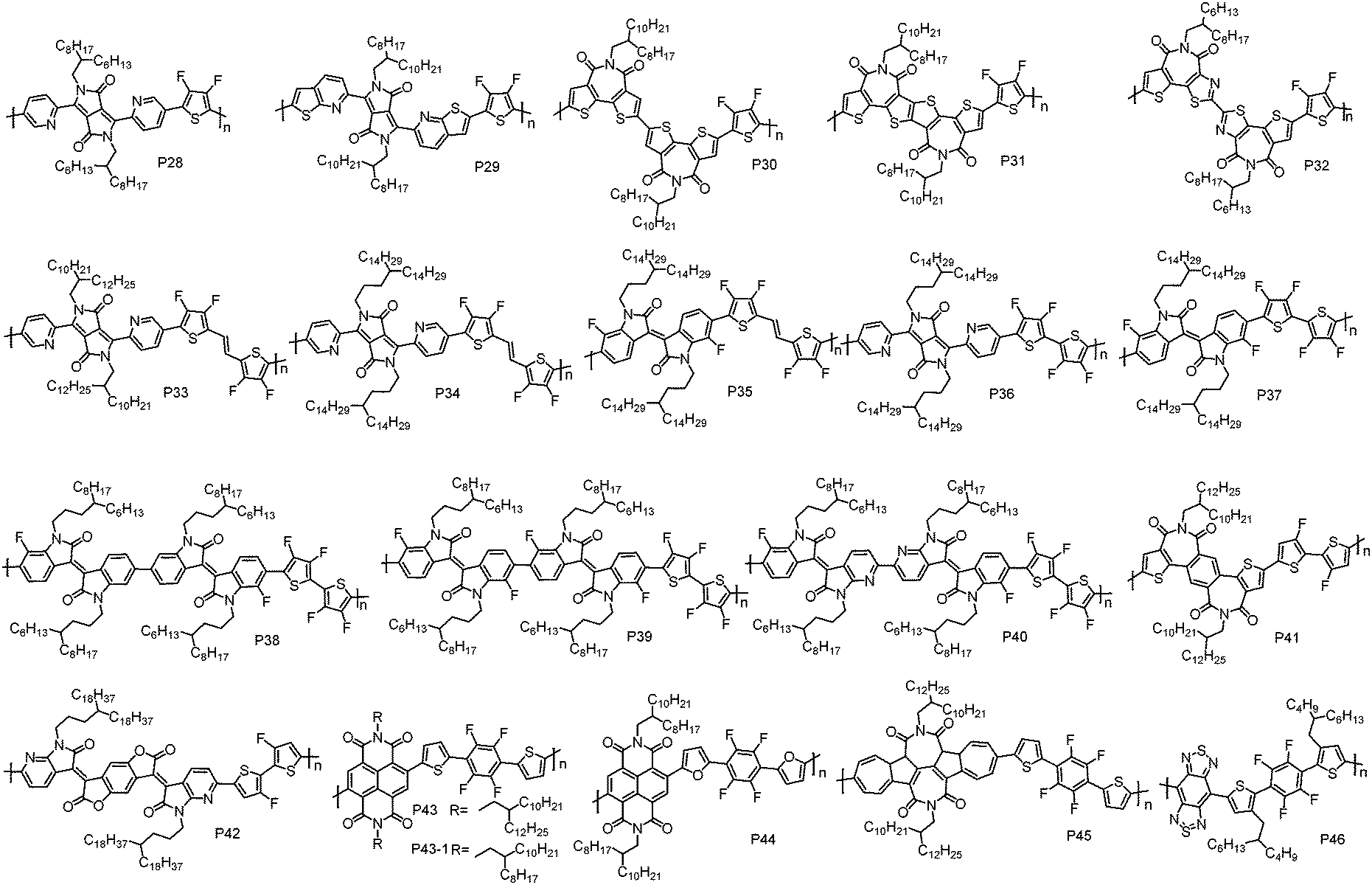 | ||
| Fig. 3 Molecular structures of the n-type D–A conjugated polymers based on F atom substituted weak D units. | ||
| Polymers | M n (kDa) | Đ | E HOMO (eV) | E LUMO (eV) | OTFT structure | μ e (cm2 V−1 s−1) | Measured environment | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Polydispersity. b HOMO energy level measured by cyclic voltammetry, unless explicitly stated. c LUMO energy level measured by cyclic voltammetry, unless explicitly stated. d Measured by photoelectron spectroscopy. e Estimated from EHOMO = ELUMO − Eoptg, in which Eoptg is the optical bandgap. f Estimated from ELUMO = EHOMO + Eoptg. | ||||||||
| P28 | 12.0 | 2.30 | −6.11 | −3.81 | BGBC | 0.11 | Nitrogen | 98 |
| P29 | 24.4 | 5.10 | −5.80d | −4.10f | TGBC | 0.10 | Nitrogen | 99 |
| P30 | 19.5 | 1.68 | −5.28e | −3.30 | TGBC | 0.82 | Nitrogen | 100 |
| P31 | 13.8 | 2.18 | −5.27e | −3.43 | TGBC | 1.13 | Nitrogen | 100 |
| P32 | 14.0 | 1.20 | −5.75e | −3.71 | TGBC | 0.91 | Nitrogen | 101 |
| P33 | 102.7 | 1.77 | −5.86 | −3.66 | TGBC | 1.19 | Air | 105 |
| P34 | 126.2 | 1.70 | −5.89 | −3.67 | TGBC | 1.35 | Air | 105 |
| P35 | 52.9 | 1.54 | −5.95 | −3.80 | TGBC | 4.97 | Air | 26 |
| P36 | 120.2 | 2.26 | −5.96 | −3.69 | TGBC | 2.45 | Air | 26 |
| P37 | 88.0 | 1.94 | −6.09 | −3.92 | TGBC | 1.35 | Air | 105 |
| P38 | 48.9 | 2.03 | −5.95 | −3.89 | TGBC | 0.93 | Air | 29 |
| P39 | 17.0 | 1.94 | −6.06 | −3.99 | TGBC | 0.71 | Air | 29 |
| P40 | 20.3 | 2.20 | −6.04 | −4.01 | TGBC | 1.24 | Air | 29 |
| P41 | 75.0 | 4.48 | −5.74 | −3.76f | BGTC | 0.40 | n/a | 106 |
| 37.0 | 3.24 | −5.72 | −3.71f | TGBC | 0.34 | Air | 107 | |
| P42 | 34.2 | 1.70 | −6.04 | −4.10 | BGTC | 0.77 | Air | 108 |
| P43 | 7.8 | 1.67 | −5.50d | −3.85 | TGBC | 1.30 | Nitrogen | 109 |
| P43-1 | 12.0 | 1.82 | −5.46e | −3.77 | TGBC | 0.16 | Nitrogen | 110 |
| P44 | 6.6 | 1.45 | −5.75d | −3.75 | TGBC | 0.40 | Nitrogen | 109 |
| P45 | 13.5 | 3.26 | −5.76 | −3.60 | BGTC | 0.42 | Nitrogen | 111 |
| P46 | 3.20 | 1.40 | −5.64 | −3.87 | BGTC | 3.70 × 10−4 | Vacuum | 112 |
Guo et al. designed and synthesized two bithiophene imide (BTI) derivatives, namely, single bond connected BTI dimers (s-2BTI) and fused BTI dimers (f-2BTI).100 The outer thiophene rings in the BTI unit could tackle the steric hindrance challenge found in NDI- and PDI-based polymers, leading to planar polymer backbones. With these two building blocks, P30 and P31 have been developed using 2FT as the comonomer. It was found that the LUMO energy level of P31 was 0.13 eV lower than that of P30, suggesting that ring fusion leads to an enhanced electron-accepting ability. Moreover, geometry optimization revealed that P31 had a more linear and planar backbone than P30, thus resulting in higher structural order with stronger interchain interactions. Therefore, relative to P30, which exhibited a μe of 0.82 cm2 V−1 s−1, P31 showed an improved μe of 1.13 cm2 V−1 s−1 in TGBC OTFT devices. Recently, a single bond connected thiazole imide dimer (DTzTI) and its copolymer with 2FT (P32) were synthesized by Guo et al.101 P32 showed lower HOMO and LUMO energy levels as compared to P30, due to the electron-deficient nature of the thiazole unit,102,103 which would help realize unipolar n-type transport. Another benefit of introducing the thiazole unit is that it leads to a more planar polymer backbone, owing to the less steric demanding I–N atom versus CH moiety in thiophene and the S⋯N intramolecular noncovalent interactions promoted by the N atom.97 TGBC devices with P32 showed pure n-type transport characteristics with the highest μe of 0.91 cm2 V−1 s−1.
TVT has been widely used to construct p-type polymers with very high mobility (>8 cm2 V−1 s−1).55,104 By introducing four F atoms on the β-positions of thiophene units in TVT, a weak D unit, (E)-1,2-bis(3,4-difluorothien-2-yl)ethene (4FTVT), was designed and synthesized by Geng et al.96 Notably, the introduction of F atoms at the β-positions remarkably enhanced the direct arylation reactivity of α C–H bonds in the thiophene unit and evaded the unwanted possible β C–H activation. Accordingly, 4FTVT exhibited high reactivity in direct arylation polycondensation (DArP). Based on 4FTVT and PyDPP units, P33 with molecular weight >100 kDa has been synthesized via DArP by using Herrmann's catalyst.105 P33 showed a deep HOMO level of −5.86 eV. DFT calculations revealed that P33 had a planar skeleton despite the existence of connecting pyridine and thiophene rings, which usually leads to a large torsion angle (∼20°) and a twisted polymer backbone. The planar backbone of P33 can be attributed to the presence of F⋯H noncovalent interactions. In TGBC OTFT devices, P33 displayed a unipolar n-type characteristic with a μe of 1.19 cm2 V−1 s−1. By moving the bifurcation of the side alkyl chains away from the polymer backbone, P34 was synthesized by Geng et al.105 P34 showed comparable HOMO and LUMO energy levels with P33, indicating that the alkyl chain branching point has a negligible influence on the energy levels. However, compared to P33, P34 showed an enhanced thin-film order and a smaller π–π stacking distance, thus a higher μe of 1.35 cm2 V−1 s−1 was achieved in P34-based TGBC OTFT devices. With 4FTVT and fluorinated isoindigo (FIID) units, another n-type polymer, P35, was synthesized by Geng et al. via DArP.26 The LUMO and HOMO energy levels for P35 are −3.80 and −5.95 eV, respectively, which are lower than those of P34. This can be explained by the multifluorination effect, that is, P35 was fluorinated on both D and A units while P34 was fluorinated only on the D unit. A unipolar high μe of 4.97 cm2 V−1 s−1 was achieved in P35-based TGBC OTFT devices. It should be noted that the OTFT devices based on P35 exhibited good air stability. No obvious degradation was observed for TGBC OTFT devices after being stored under air conditions for three months, and the μe value was still as high as 0.05 cm2 V−1 s−1 after two month storage in air for BGTC OTFT devices without any encapsulation. The good air stability can be attributed to the low-lying LUMO energy level of P35, which can resist the electron capture by water and oxygen during the device operation in air. Additionally, the hydrophobic nature of P35 caused by the multifluorination can also prevent the penetration of oxygen and water into the polymer thin-film.
Although P33, P34 and P35 showed unipolar n-type characteristics, an increase of the current was found in their transfer characteristics when the VGS was below the turn-on voltage, indicating that weak hole transport still existed in the OTFT devices. It suggests that the HOMO energy levels of P33, P34 and P35 are not low enough to completely block the hole injection. As we discussed above, the existence of a vinylene unit would elevate the HOMO energy level. In order to obtain strictly unipolar n-type characteristics, another weak D unit, 3,3′,4,4′-tetrafluoro-2,2′-bithiophene (4FBT), was synthesized by Geng et al. Like 4FTVT, 4FBT also showed high reactivity in DArP, and high molecular weight CPs without defects can also be synthesized via DArP. With 4FBT as the comonomer, P36 and P37 have been developed via DArP using PyDPP and FIID as the A units, respectively.26,105 Compared to the 4FTVT-based analogues, 4FBT-based polymers indeed showed lower HOMO energy levels, which are −5.96 and −6.09 eV for P36 and P37, respectively. As expected, P36 and P37 exhibited purely unipolar electron transport behaviour in TGBC OTFT devices, reflected by their typical unipolar n-type output characteristics and clear off-regimes in their transfer characteristics. The μe values for P36 and P37 were 2.45 and 1.35 cm2 V−1 s−1, respectively. Recently, a series of n-type CPs P38, P39 and P40 were developed by Geng et al. based on 4FBT and bisisoindigo (bis-IID) derivatives containing different numbers of F or I–N atoms.29 All these polymers showed low-lying LUMO and HOMO energy levels, and their energy levels can be feasibly tuned by the numbers of F and I–N atoms. TGBC OTFT devices based on these polymers all displayed unipolar n-type transport behaviours. Among these polymers, P40 exhibited the highest μe of 1.24 cm2 V−1 s−1.
3,3′-Difluoro-2,2′-bithiophene (2FBT) and 1,2,4,5-tetrafluorobenzene (TFB) have also been used as the weak D units to construct n-type polymer. P41 comprising 2FBT and dithienylbenzodiimide (TBDI) was developed by Guo et al.106 Unipolar n-type charge transport properties with the highest μe of 0.40 cm2 V−1 s−1 were achieved in P41-based BGTC OTFT devices. Soon after, Liao et al. also reported the synthesis of P41, and they found that P41 exhibited a similar μe of 0.34 cm2 V−1 s−1 in TGBC OTFT devices.107 Another example of an n-type polymer with 2FBT as the weak D unit is P42, which was developed by Zhang et al.108 The BGTC OTFT devices with P42 showed unipolar electron transport characteristics with a high μe of 0.77 cm2 V−1 s−1. However, obviously an undesirable kink was observed in the |ISD|1/2–VGS curves, which led to the overestimation of mobility. Therefore, the effective mobility was calculated using the reliability factor (r ≈ 30%), which was 0.23 cm2 V−1 s−1. Notably, P42 exhibited excellent air stability of electron transport. For BGTC OTFT devices of P42 without any encapsulation, the unipolar electron mobility was still as high as 0.10 cm2 V−1 s−1 with on/off ratios of >106 after 60 days of air storage.
Like 4FTVT and 4FBT, TFB also showed high reactivity in DArP. With TFB and NDI units, P43 and P44 were synthesized by Sommer et al. via DArP.109 The linkage unit, thiophene in P43 and furan in P44, significantly reduced the steric hindrance between NDI and TFB units due to the weak interactions of F⋯S/F⋯O and the avoidance of directly connecting benzene rings. Due to the presence of a debromination side reaction, Mns of the polymers are relatively low, which are 7.8 kDa for P43 and 6.6 kDa for P44. Compared to P44, which exhibited a μe of 0.40 cm2 V−1 s−1, P43 showed a more efficient electron transport with the highest μe of up to 1.30 cm2 V−1 s−1. The enhanced OTFT performance of P43 can be explained by the reduced orientational disorder and the formation of a terrace-like thin-film morphology. Recently, P43-1 with a Mn of 12 kDa was developed by Jin et al. via Stille coupling polycondensation.110 In TGBC OTFT devices, P43-1 displayed a unipolar μe of 0.16 cm2 V−1 s−1, which is almost one order of magnitude lower than that of P43. The dramatically decreased electron transport performance of P43-1 can be attributed to its disordered thin-film. As mirrored by the XRD analysis, up to 5 orders of (h00) diffraction peaks were observed in the P43 thin-film, while P43-1 showed an almost amorphous feature. This can be ascribed to the poor solubility of P43-1 due to the small size of side alkyl chains, leading to the difficulty in forming smooth thin-films. Other than NDI, rylene diimide with an azulene unit was recently reported to be another promising candidate for constructing n-type CPs. Gao et al. synthesized P45 by copolymerizing TFB with 2,2′-biazulene-1,1′,3,3′-tetracarboxylic diimides (TBAzDI).111 In BGTC OTFT devices, P45 showed a μe of 0.42 cm2 V−1 s−1. Very recently, Wang et al. reported a CP comprising TFB and benzobisthiadiazole, namely, P46.112 Relative to its non-fluorinated counterpart that showed a p-type charge transport characteristic in BGTC OTFT devices,112 P46 exhibited unipolar n-type charge transport behaviour due to its deep HOMO energy level, which could build large hole injection barriers. This result again highlights the advantage of weak D units for constructing n-type CPs.
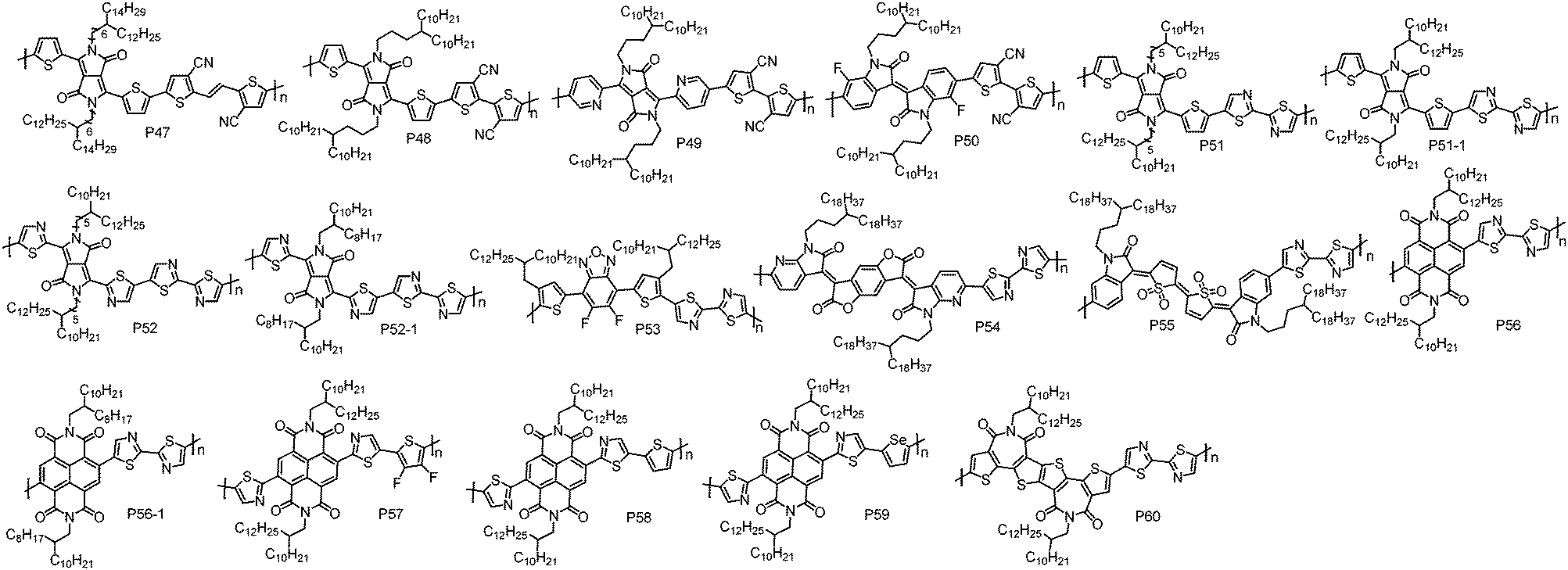 | ||
| Fig. 4 Molecular structures of the n-type D–A conjugated polymers based on CN groups and I–N substituted weak donor units. | ||
| Polymers | M n (kDa) | Đ | E HOMO (eV) | E LUMO (eV) | OTFT structure | μ e (cm2 V−1 s−1) | Measured environment | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Polydispersity. b HOMO energy level measured by cyclic voltammetry, unless explicitly stated. c LUMO energy level measured by cyclic voltammetry, unless explicitly stated. d Measured by photoelectron spectroscopy. e Estimated from ELUMO = EHOMO + Eoptg. f Measured by photoemission yield spectroscopy. g Measured by low-energy inverse photoemission spectroscopy. h With calcium and ethoxylated polyethylenimine (PEIE) modified silver as source and drain electrodes. i The device was fabricated by blade-coating. j The device was fabricated by off-centre spin-coating. | ||||||||
| P47 | 53.0 | 1.30 | −5.40 | −4.10e | TGBC | 1.20 | Nitrogen | 113 |
| P48 | 155.0 | 1.34 | −5.41 | −3.67 | TGBC | 0.35 | Air | 27 |
| P49 | 275.0 | 1.30 | −5.83 | −3.75 | TGBC | 0.30 | Air | 27 |
| P50 | 111.0 | 1.54 | −6.15 | −3.92 | TGBC | 0.25 | Air | 27 |
| P51 | 64.0 | 3.60 | −5.54d | −3.75 | BGTC | 0.31h | Nitrogen | 115 |
| P51-1 | 18.0 | 3.80 | −5.56 | −3.63 | TGBC | 0.53 | Air | 116 |
| P52 | 26.0 | 1.35 | −5.71d | −4.10e | BGBC | 0.067 | Nitrogen | 37 |
| P52-1 | 44.0 | 1.74 | −5.92 | −4.32e | BGTC | 0.041 | Air | 118 |
| P53 | 31.9 | 1.70 | −5.87 | −4.13e | TGBC | 0.83 | Air | 119 |
| P54 | 40.4 | 2.58 | −6.06 | −4.28 | BGTC | 0.055 | Vacuum | 120 |
| P55 | 14.3 | 2.50 | −5.99 | −4.18 | BGBC | 0.017 | Nitrogen | 95 |
| P56 | 45.0 | 3.50 | −5.78d | −4.40 | BGBC | 0.05i | Nitrogen | 121 |
| P56-1 | 89.0 | 4.98 | n/a | −3.83 | TGBC | 0.085j | Nitrogen | 122 |
| P57 | 79.4 | 1.80 | −6.24 | −3.89 | TGBC | 0.57 | Air | 28 |
| P58 | 115.6 | 1.80 | −6.01 | −3.85 | TGBC | 0.37 | Air | 28 |
| P59 | 118.3 | 1.50 | −5.91 | −3.83 | TGBC | 0.39 | Air | 28 |
| P60 | 56.1 | 2.03 | −6.12f | −3.56g | BGTC | 0.05 | Air | 123 |
With bithiazole and thiophene flanked difluorobenzoxadiazole, P53 was developed by Guo et al.119 Notably, although electron-rich thiophene units were flanked on the difluorobenzoxadiazole unit, P53 had a low-lying HOMO energy level of −5.87 eV, which could block hole injection. P53-based TGBC OTFT devices with CYTOP as the dielectric showed electron transport behaviour with a μe of 0.71 cm2 V−1 s−1. During device optimization, they found that the devices with PMMA dielectric displayed an improved performance with a higher μe of up to 0.83 cm2 V−1 s−1. Subsequently, Cho et al. reported P54 based on bithiazole and a BDOPV derivative.120 P54 showed deep LUMO/HOMO energy levels of −4.28/−6.06 eV, which were low enough to stabilize electron transport in air and to block hole injection. Consequently, P54 BGTC OTFT devices without any encapsulation exhibited unipolar electron transport with a μe of 0.055 cm2 V−1 s−1 under air conditions. Importantly, the BGTC OTFT devices displayed excellent air stability and maintained unipolar electron transport with a μe of up to 0.01 cm2 V−1 s−1 after one year storage in air. The copolymer (P55) of quinoidal acceptor IDTO and a bithiazole unit was synthesized by Li et al.95 In BGBC OTFT devices, P55 exhibited inferior performance with a μe of 0.017 cm2 V−1 s−1 due to its low molecular weight and the amorphous thin-film.
As aforementioned, the NDI unit has been widely used to construct n-type CPs. However, weak hole injection can be observed for the most of NDI-based CPs due to their rather high HOMO energy levels.28 To address this issue, Reichmanis et al. synthesized a n-type polymer P56 containing bithiazole and NDI units.121 As expected, P56 showed ∼0.4 eV deeper HOMO energy level than the NDI–bithiophene copolymer (N2200). Therefore, P56 exhibited a strictly unipolar n-type transport characteristic. The BGBC OTFT devices of P56 fabricated by blade-coating displayed a μe of 0.05 cm2 V−1 s−1, which is lower than that of N2200. The low μe of P56 may be correlated with its twisted conjugated backbones derived from the steric hindrance between NDI and thiazole units, hindering polymer backbone organization into highly ordered microstructures that would be expected to result in efficient intermolecular charge transport. A similar polymer P56-1 was synthesized by Sommer et al. via DArP with Pd2dba3 and tris(o-anisyl)phosphine as the catalyst and ligand, respectively.122 It should be noted that homo-coupling free P56-1 with a high molecular weight of up to 89.0 kDa can be synthesized by DArP in less than 1 hour, suggesting that DArP is an efficient protocol to synthesize CPs. P56-1 showed unipolar electron transport with the highest μe of 0.085 cm2 V−1 s−1 in TGBC OTFT devices fabricated by the off-centre spin-coating method. In order to simultaneously reduce the HOMO energy level and improve the planarity of polymer skeletons, Cao et al. designed and synthesized a thiazole flanked NDI unit (TzNDI) with the thiazole N atom pointing toward the NDI unit.28 With this linking method, the twist angle between NDI and thiazole could be significantly reduced due to the less steric demanding I–N. Based on TzNDI, a series of n-type polymers have been developed, including P57, P58 and P59. Indeed, the TzNDI unit endows the resultant polymers with good backbone planarity and low-lying HOMO energy levels. In TGBC OTFT devices, all these polymers exhibited strictly unipolar electron transport performance with the highest μe of up to 0.57 cm2 V−1 s−1, outperforming N2200 (0.41 cm2 V−1 s−1) under the same device fabrication conditions.
f-2BTI with the imide groups located at the centre of bithiophene should minimize the steric hindrance between the carbonyl bridge and the adjacent atoms to achieve a planar polymer backbone. By copolymerizing f-2BTI and a bithiazole unit, Takimiya et al. synthesized P60.123 The polymer showed a deep-lying HOMO energy level of −6.12 eV as determined by photoemission yield spectroscopy (PYS). In OTFT devices with the BGTC configuration, P60 exhibited a unipolar μe of 0.05 cm2 V−1 s−1.
3. D–A CPs with dual-A
Introducing two electron acceptor building blocks into a polymer backbone to construct dual-A architecture (A1–D–A2–D) was an effective approach to tune the energy levels of D–A polymers.124–126 This strategy could increase the loading of A unit in polymer chains, enabling the ease of realizing unipolar n-type transport via further energy level modulation (Fig. 5 and Table 5).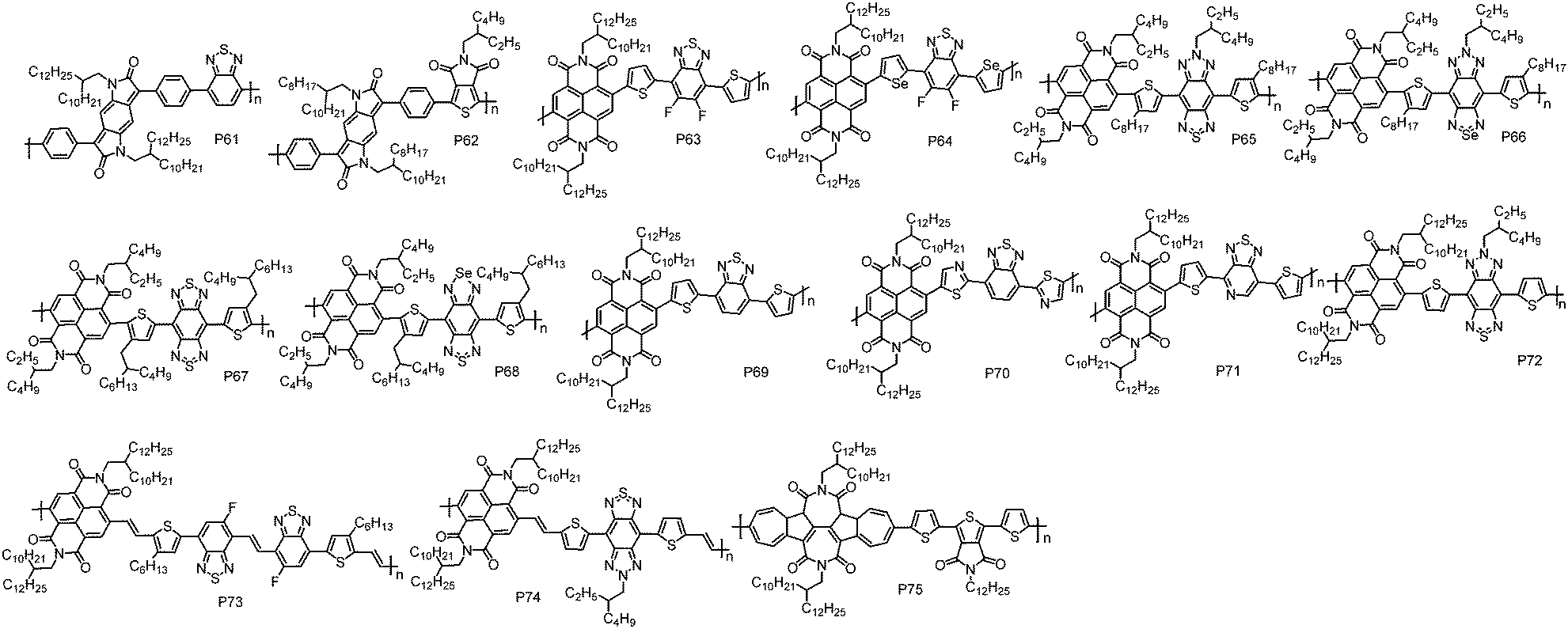 | ||
| Fig. 5 Molecular structures of the n-type D–A conjugated polymers based on dual-A. | ||
| Polymers | M n (kDa) | Đ | E HOMO (eV) | E LUMO (eV) | OTFT structure | μ e (cm2 V−1 s−1) | Measured environment | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Polydispersity. b HOMO energy level measured by cyclic voltammetry, unless explicitly stated. c LUMO energy level measured by cyclic voltammetry, unless explicitly stated. d Measured by photoelectron spectroscopy. e Estimated from ELUMO = EHOMO + Eoptg. f The Au electrodes were modified with PFBT. g The device was modified with NTMS. | ||||||||
| P61 | 54.0 | 1.25 | −5.84d | −3.99e | TGBC | 0.01 | n/a | 127 |
| P62 | 147.0 | 2.65 | −5.98d | −4.22e | BGTC | 1.3 × 10−3 | n/a | 128 |
| P63 | 48.5 | 4.56 | −6.24 | −3.85 | TGBC | 2.2f | Air | 129 |
| P64 | 56.1 | 4.24 | −6.20 | −3.88 | TGBC | 3.5f | Air | 129 |
| P65 | 11.1 | 1.84 | −5.35 | −3.72 | BGTC | 1.1 × 10−3 | Vacuum | 133 |
| P66 | 10.2 | 1.53 | −5.20 | −3.85 | BGTC | 4.1 × 10−3 | Vacuum | 133 |
| P67 | 11.9 | 1.54 | −5.68 | −3.93 | BGTC | 0.039 | Vacuum | 133 |
| P68 | 10.3 | 1.61 | −5.34 | −4.03 | BGTC | 2.7 × 10−3 | Vacuum | 133 |
| P69 | 313.6 | 3.40 | −5.77 | −3.78 | BGTC | 0.92 | Vacuum | 134 |
| P70 | 122.5 | 2.70 | −6.01 | −3.86 | BGTC | 0.46 | Vacuum | 134 |
| P71 | 141.0 | 2.00 | −5.87 | −3.81 | BGTC | 2.11 | Vacuum | 134 |
| P72 | 133.1 | 2.50 | −5.45 | −3.88 | BGTC | 5.35g | Vacuum | 134 |
| P73 | 31.6 | 2.50 | −5.71 | −3.80 | BGTC | 3.87g | Vacuum | 45 |
| P74 | 54.9 | 1.80 | −5.40 | −3.87 | BGTC | 7.16g | Vacuum | 45 |
| P75 | 22.1 | 3.68 | −5.82 | −3.76 | BGTC | 0.42 | Nitrogen | 111 |
Benzodipyrrolidone (BDP), which is similar in structure to the DPP unit, is a promising building block for CPs with high mobility. McCulloch et al. reported an A1–D–A2–D polymer P61 by the combination of BDP with a benzothiadiazole (BT) unit.127 Compared with the typical D–A polymers with BDP as the A unit,127 P61 indeed showed a much lower HOMO energy level of −5.84 eV, being suitably deep to block hole injection from the commonly used Au electrodes. In TGBC OTFT devices, P61 displayed electron transport with a μe of 0.01 cm2 V−1 s−1. Afterward, another A1–D–A2–D polymer P62 based on BDP and thienopyrroledione (TPD) was synthesized by Kanbara et al. via DArP with Pd(AcO)2 and PCy3·HBF4 as the catalyst and ligand, respectively.128 However, P62 exhibited inferior electron transport performance with a μe of 0.0013 cm2 V−1 s−1, due to its amorphous thin-film likely originating from the steric hindrance of the bulky alkyl chains.
NDI was often selected as one of the A moieties in dual-A polymers due to its strong electron-withdrawing nature. Based on NDI and fluorinated benzothiadiazole (FBTz), P63 with a A1–D–A2–D structure was synthesized by Liu et al.129 Using thiophene as the linkage could avoid the steric hindrance between NDI and FBTz, and thus maintain good planarity of the polymer backbone. Cyclic voltammetry experiments revealed that P63 exhibited a strong reduction process and a weak oxidation process with the LUMO/HOMO energy levels of −3.85 eV/−6.24 eV. P63 showed unipolar electron transport with the highest μe of 2.20 cm2 V−1 s−1 in TGBC OTFT devices, where pentafluorobenzenethiol (PFBT) modified Au was used as the source and drain electrodes to facilitate the electron injection. It has been demonstrated that the work function of Au electrodes was tuned from 5.13 eV (bare Au) to 4.77 eV (PFBT modified Au),130 and therefore the energetic mismatch between Au electrodes and the LUMO energy level of the polymer was reduced. Compared with sulfur, selenium has a larger atomic radius, which could facilitate orbital overlap and charge carrier transport. Besides, the empty orbital in selenium could improve the electron-accepting ability of selenophene, and in turn decrease the LUMO energy level of selenophene-containing polymers.131,132 With these advantages, P64 was developed by Liu et al., in which selenophene was used as the linking unit.129 It was found that P64 possessed a narrower bandgap and deeper LUMO energy level than P63. Thanks to the smooth surface morphology and short π–π stacking distance, P64 exhibited a high μe of 3.50 cm2 V−1 s−1 in TGBC OTFT devices.
Soon after, Michinobu et al. reported a series of A1–D–A2–D polymers, namely, P65, P66, P67 and P68, composed of NDI and benzobisthiadiazole (BBT) or its heteroatom-substituted derivatives (SN).133 Similar to Liu's results, replacing the sulfur atom in the thiadiazole heterocycles with a selenium atom led to deeper LUMO energy levels and stronger π–π interactions of the polymers. Consequently, P66 (0.0041 cm2 V−1 s−1) exhibited three times higher μe than P65 (0.0011 cm2 V−1 s−1) at the optimized annealing temperature of 250 °C in BGTC OTFT devices. However, the other selenium containing polymer, P68 (0.0027 cm2 V−1 s−1), showed much lower μe than its thiophene containing analogue P67 (0.039 cm2 V−1 s−1) due to its low thermal stability and decomposition problem during the thermal annealing process. Subsequently, Michinobu et al. developed another four A1–D–A2–D polymers, P69, P70, P71 and P72, based on NDI and BT units.134 With these polymers, the effect of different N-substituted positions on the OTFT device performance was studied in detail. It should be noted that an optimized Stille polymerization protocol with chlorobenzene as the solvent and Pd(0)/Cu(I) as the cocatalyst was developed to synthesize high molecular weight polymers. Chlorobenzene provides good solubility for these polymers, while Cu(I) can react with organostannanes to produce transient organocopper intermediates and thereby accelerate the transmetallation reaction in the catalytic circle,135 thus giving rise to high molecular weight polymers. BGTC OTFT devices with P69 showed electron dominant transport properties with a μe of 0.92 cm2 V−1 s−1. Replacing the thiophene unit in P69 with thiazole produced P70, which showed a unipolar electron transport characteristic with a μe of 0.46 cm2 V−1 s−1. P71, which contained I–N on the electron-withdrawing BT unit, exhibited an even higher μe of 2.11 cm2 V−1 s−1. P72 with two I–N atoms embedding into the triple fused ring structure of the BT unit displayed the highest μe of 4.87 cm2 V−1 s−1. However, due to the high HOMO energy level of P72, weak hole transport was observed in the OTFT devices. In order to realize unipolar electron transport properties, the BGTC OTFT devices based on P72 was modified with [3-(N,N-dimethylamino)-propyl]trimethoxysilane (NTMS), in which the amine groups could trap the hole and thus suppress the hole transport. In such a case, a unipolar μe of up to 5.35 cm2 V−1 s−1 was obtained. P65 and P72 possessed similar polymer backbones and only differed in the alkyl chains. P65 contained small alkyl chains on both D and A units while P72 carried long alkyl chains on the A unit. However, the μe of P72 is three orders of magnitude higher than that of P65. This result suggests that the position and selection of alkyl chains are as important as optimizing the polymer backbone, since the alkyl chains not only affect the solubility of the polymer but also influence the packing behaviour of the polymer and therefore the device performance.136,137
As we discussed above, the connection of NDI and a thiophene unit (NDT–T) would create large torsion and lead to a twisted polymer backbone, which impairs charge transport. Inserting a vinylene group between NDI and the thiophene unit (NDI–V–T) provides an effective way to solve this issue, due to the formation of H⋯O intramolecular hydrogen bonds between vinylene and NDI units, ensuring planar backbones of the polymer.97 With this design strategy, P73 and P74 were recently developed by Michinobu et al.45 In P73, the F atom on the BT unit can form additional intramolecular hydrogen bonds via H⋯F interactions, further locking the conformation of polymer backbones. As demonstrated by the DFT calculations, the torsion angles were significantly decreased after incorporating vinylene groups and F atoms, thus planar backbones were obtained in P73 and P74, which in turn led to high order polymer thin-films. In BGTC OTFT devices modified with NTMS, P73 displayed unipolar electron transport with a high μe of 3.87 cm2 V−1 s−1. P74 exhibited a much higher μe of 7.16 cm2 V−1 s−1, which is the highest value for unipolar n-type CPs. 2D-GIWAXS revealed that P73 adopted an edge-on orientation molecular packing with a π–π stacking distance of 3.49 Å, while P74 showed a bimodal packing behaviour with a mixture of face-on and edge-on backbone orientations and an extremely short π–π stacking distance of 3.40 Å. It is believed that the bimodal packing is promising for achieving high mobility because the face-on orientation packing molecules can provide a chance for charge carriers to move around the boundaries between crystalline and amorphous domains.138–140 Moreover, compared with edge-on orientation, face-on molecular packing is beneficial for the injection of charge carriers from electrodes to the semiconductor layer.141 These events were facilitated by the extremely short π–π stacking distance. Therefore, better electron transport was realized for P74 in comparison with P73.
TBAzDI, an imide-functionalized azulene acceptor unit, was considered as another promising building block for n-type CPs. Recently, an A1–D–A2–D polymer P75 was developed by Gao et al.111 DFT calculations revealed that the LUMO orbital of P75 was well delocalized over the entire polymer backbone, which was greatly different from those of NDI- and PDI-based n-type D–A polymers, where the LUMO orbitals were usually localized on the A units.73,142 The delocalized LUMO orbital could encourage LUMO–LUMO interactions, improving electron transfer between polymer chains. The LUMO and HOMO energy levels of P75 are −3.76 and −5.82 eV, respectively, lower than those of TBAzDI-based polymer with a weak D unit of TFB (P45). The above results indicate that the dual-A design strategy not only significantly brings down the energy levels of polymers but also promotes the delocalization of LUMO orbitals, which are of critical significance for n-type polymers. In BGTC OTFT devices, P75 exhibited a high μe of 0.42 cm2 V−1 s−1, making P75 among the best n-type CPs for BGTC OTFT devices.
4. Acceptor–acceptor (A–A) type CPs
Recently, a new design concept, called “A–A” type polymer, has been proposed to create n-type CPs. This strategy can eliminate the adverse effect from the electron-rich D unit completely, yielding unipolar n-type CPs feasibly. However, the synthesis of A–A type CPs with high molecular weights, which dramatically affects the charge transport performance of CPs,143,144 remains a great challenge due to: (1) the difficulty in synthesizing high purity organometal functionalized A monomers because of their reduced chemical reactivity; (2) the instability nature of the organometal functionalized electron-accepting heteroarenes.145 Until now, only a few A–A type CPs have been reported, and they are commonly synthesized via Yamamoto146–148 or Stille polycondensation.101,149–151 Certainly, some A–A type CPs, such as the polymers comprising a IID or BT unit, also can be synthesized by Suzuki polycondensation owing to the accessibility and stability of their organoboron derivatives.152–155 Notably, the recent advances in DArP have significantly promoted the development of this class of polymers.156,157 From the perspective of material design, A–A type CPs are prone to suffer from the problem of twisted polymer backbones because the electron-withdrawing substituents/groups, such as imide and cyano groups that can endow the A units with low-lying frontier molecular orbital energy levels, are usually bulky and induce significant steric crowding between the adjacent A units.45,149 The twisted polymer chains would cause poor molecular packing and less ordered thin-films, impairing charge transport. Therefore, in order to reduce the backbone torsion of A–A type CPs, elaborate molecular design is required (Fig. 6 and Table 6).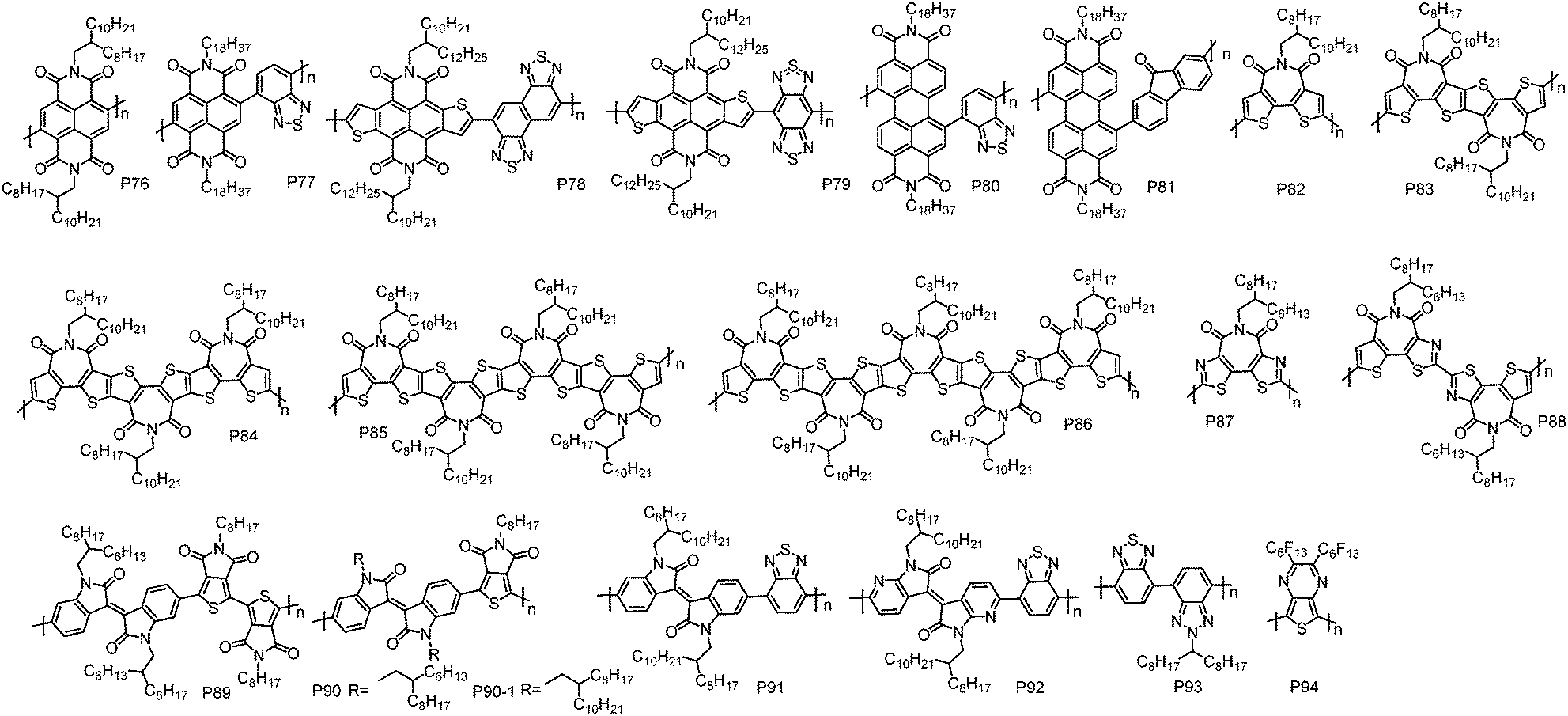 | ||
| Fig. 6 Molecular structures of the n-type A–A conjugated polymers. | ||
| Polymers | M n (kDa) | Đ | E HOMO (eV) | E LUMO (eV) | OTFT structure | μ e (cm2 V−1 s−1) | Measured environment | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Polydispersity. b HOMO energy level measured by cyclic voltammetry, unless explicitly stated. c LUMO energy level measured by cyclic voltammetry, unless explicitly stated. d Measured by photoelectron spectroscopy. e Estimated from EHOMO = ELUMO − Eoptg, in which Eoptg is the optical bandgap. f Estimated from ELUMO = EHOMO + Eoptg. g Estimated from TGBC OTFT devices with polyolefin–polyacrylate dielectric. h The device was modified with CsF. i The device was fabricated by off-centre spin coating. | ||||||||
| P76 | 71.1 | 2.90 | −6.50e | −3.76 | BGTC | 6.0 × 10−4 | Nitrogen | 146 |
| P77 | 5.2 | 1.74 | −5.14 | −3.03 | BGTC | 0.20 | n/a | 152 |
| P78 | 15.7 | 1.73 | −6.20d | −4.20 | BGTC | 0.21 | Air | 159 |
| P79 | 20.5 | 2.53 | −5.50 | −4.40 | BGTC | 0.31 | Air | 160 |
| P80 | 30.6 | 1.16 | −5.49 | −3.08 | BGTC | 0.032 | n/a | 152 |
| P81 | 8.2 | 1.38 | −5.94 | −4.00 | BGTC | 0.01 | Air | 161 |
| P82-L | 3.6 | 2.20 | −6.28 | −3.47 | BGTC | 0.011 | Vacuum | 147 |
| P82-M | 7.2 | 1.98 | −6.28 | −3.47 | BGTC | 0.038, 0.14g | Air, vacuum | 148 |
| P82-M* | 5.4 | 1.43 | n/a | n/a | n/a | 0.018h | Nitrogen | 149 |
| P82-H | 12.7 | 2.12 | −5.46e | −3.48 | TGBC | 1.53,h 3.71h,i | Nitrogen | 149 |
| P83 | 13.3 | 2.20 | −5.39e | −3.53 | TGBC | 1.34h | Nitrogen | 149 |
| P84 | 11.7 | 1.90 | −5.41e | −3.59 | TGBC | 0.53h | Nitrogen | 149 |
| P85 | 9.4 | 1.60 | −5.47e | −3.67 | TGBC | 0.21h | Nitrogen | 149 |
| P86 | 9.2 | 1.10 | −5.49e | −3.72 | TGBC | 0.014h | Nitrogen | 149 |
| P87 | 5.8 | 2.60 | −6.17e | −3.94 | TGBC | 0.015i | Nitrogen | 101 |
| P88 | 7.0 | 1.10 | −5.78e | −3.77 | TGBC | 1.61i | Nitrogen | 163 |
| P89 | 20 | 2.20 | −6.10 | −4.20 | BGBC | 3.5 × 10−3 | Nitrogen | 156 |
| P90 | 24 | 2.20 | −6.00 | −4.20 | BGBC | 3.0 × 10−4 | Nitrogen | 156 |
| P90-1 | 57.1 | 3.30 | −5.70 | −3.66 | BGTC | 0.01 | Nitrogen | 153 |
| P91 | 25.0 | 1.32 | −5.68 | −3.54 | BGTC | 0.22 | Nitrogen | 153 |
| P92 | 14.0 | 1.60 | −5.80d | −4.10f | TGBC | 1.0 | n/a | 154 |
| P93 | 120 | 1.50 | −5.40d | −3.40f | TGBC | 0.020 | Nitrogen | 145 |
| P94 | n/a | n/a | n/a | −4.12 | TGBC | 2.2 × 10−6 | Air-free | 151 |
The promising electron transport properties of NDI-based polymers inspire the exploration of A–A type polymers with an NDI unit. An NDI-based homopolymer P76 with branched 2-octyldodecyl substituents was first synthesized by Luscombe et al. via Yamamoto coupling polycondensation.146 Compared with the NDI–thiophene D–A polymers, P76 showed much lower HOMO energy level, which is −6.50 eV, suggesting that A–A type CPs are highly desirable as unipolar electron transport semiconductors. However, P76 exhibited a limited μe of 6.0 × 10−4 cm2 V−1 s−1 due to its amorphous thin-film, which was probably derived from the steric repulsion of the NDI unit and the absence of space for alkyl chain interdigitation. Later, an NDI- and BT-based A–A type polymer P77 was developed by Iyer et al. via Suzuki polycondensation.152 As a spacer, the BT unit could avoid the steric hindrance of the NDI unit and encourage alkyl chain interdigitation, thus promoting polymer chain packing and the formation of lamellar nanostructures with compact alkyl chain interdigitation. As a result, P77 displayed highly ordered thin-films as revealed by the obvious diffraction peaks in the out-of-plane XRD. The well-organized microstructures of P77 enabled more efficient electron transport than P76. In BGTC OTFT devices, P77 showed the highest μe of 0.20 cm2 V−1 s−1.
A thiophene fused NDI derivative, 4,5,9,10-naphtho[2,3-b:6,7-b′]dithiophenediimide (NDTI), was reported by Takimiya et al.158 It is an attractive archetypical building block for CPs for the following reasons: (1) the planar and rigid structure is beneficial to charge transport; (2) the outer thiophene rings help create polymers with good planarity. A–A type polymers P78 and P79 were synthesized via CuI assisted Stille coupling polycondensation by copolymerizing NDTI with naphthobisthiadiazole (NTz) and benzobisthiadiazole (BBT), respectively.159,160 Both polymers showed near-infrared absorption spectra and remarkable low-lying LUMO energy levels of ∼−4.40 eV. As expected, both polymers afforded air stable unipolar electron transport with μe of 0.21 and 0.31 cm2 V−1 s−1 for P78 and P79, respectively, in BGTC OTFT devices.
A PDI unit has also been used as an A unit to develop A–A type polymers with unipolar electron transport characteristics. P80152 and P81161 were synthesized by Iyer et al. and Zhan et al., respectively. The μe for P80 and P81 were 0.032 and 0.01 cm2 V−1 s−1, respectively.
A BTI unit was designed and synthesized by Marks et al. in 2008.147 Moving the imide group to the centre of bithiophene could greatly attenuate the steric hindrance, therefore improving the backbone planarity of the resultant polymers. A BTI homopolymer P82 was synthesized using Yamamoto coupling polycondensation. When the reaction was conducted at 60 °C in DMF, a polymer with a low number-average molecular weight (Mn) of 3.6 kDa was obtained (P82-L), after being purified by multiple precipitations.147 By increasing the polymerization temperature from 60 °C to 80 °C, P82 with an increased Mn of 7.2 kDa (P82-M) was yielded.148 Polymerization at 80 °C using DMF and toluene as the co-solvent led to insoluble P82.148 In BGTC OTFT devices, P82-M showed a μe of 0.038 cm2 V−1 s−1, two times higher than that of P82-L (0.011 cm2 V−1 s−1). This trend is common for CPs and in good agreement with the XRD and atomic force microscopy (AFM) results, where more ordered thin-film and better-defined grains were observed for P82-M. TGBC OTFT devices with polyolefin–polyacrylate dielectric afforded an improved μe of 0.14 cm2 V−1 s−1 using P82-M as the channel material.148
Stille coupling polycondensation using hexamethylditin is another protocol to synthesize A–A type polymers. In order to investigate the effect of the polymerization method on the molecular weight, polymer structure and device performance, P82 was prepared by Stille and Yamamoto coupling polycondensation.149 Compared with Yamamoto polycondensation, which yielded P82 with a Mn of 5.4 kDa (P82-M*), Stille polycondensation furnished P82 with a high Mn of 12.7 kDa (P82-H), indicating that Stille polycondensation is a more effective method to prepare high Mn A–A type polymers. P82-H showed much better performance with the highest μe of up to 1.53 cm2 V−1 s−1 in TGBC OTFT devices modified with CsF for reducing the electron injection barriers and suppressing the weak hole injection. In contrast, P82-M* displayed a low μe of 0.018 cm2 V−1 s−1 under the same device configuration. Apart from Mn, the structural defects and Br end groups that act as charge carrier traps may also contribute to the poor performance of P82-M*. This result clearly indicates that the polymerization method and hence polymer quality are critical to the device performance. By using the off-centre spin-coating technique, the μe of P82-H can be further improved to 3.71 cm2 V−1 s−1, which is the highest reported value for A–A type n-type polymers.
With a combination of Yamamoto coupling, ester hydrolysis, carboxylic acid dehydration, and immidization reactions, a series of highly electron-deficient (semi)ladder BTI derivatives (BTIn) with up to 5 imide groups and 15 rings in a row have been synthesized by Guo et al.162 Based on these units, homopolymers PBTIn, namely, P83, P84, P85 and P86, have been developed using Stille polycondensation.149 Compared with P82, PBTIn showed red-shifted absorption and gradually down-shifted HOMO and LUMO energy levels. However, the electron transport performance of PBTIn was lower than that of P82, and a monotonic decrease of μe from 1.34 cm2 V−1 s−1 for P83 to 0.014 cm2 V−1 s−1 for P86 was observed with an extension of the conjugation length of the monomer. It is likely because that the larger π-conjugation monomer causes a reduced freedom of molecular motion in the solid state, impacting self-organization of polymer chains into ordered microstructures.
Although BTI-based A–A type polymers delivered good electron transport properties, weak hole injection could be observed in the OTFT devices without CsF modification, as indicated by the lack of clear off-regime in their transfer curves. This phenomenon could be attributed to the electron-rich thiophene unit in the BTI core, resulting in relatively high-lying HOMO energy levels of the resultant polymers. In order to bring down the HOMO energy level of the polymer and hence block hole injection, bithiazole imide (BTzI) was designed and synthesized by Guo et al. and its homopolymer P87 was developed by Stille polycondensation.101 It should be noted that the polymerization of the BTzI monomer appeared to be problematic under typical Pd-mediated Stille coupling conditions, possibly due to the chelation of the Pd catalyst with the N atoms on thiazole moieties. After adding CuI as the co-catalyst, P87 with a Mn of 5.8 kDa was obtained. As expected, P87 showed a deep HOMO energy level of −6.17 eV, which was ∼0.7 eV lower than that of P82-H when measured by the same method. However, it was found that the thin-film order of the polymer was significantly reduced after replacing the two thiophene units in the BTI core with thiazole moieties. Thus P87 showed inferior electron mobility. The highest unipolar μe of P87 is 0.015 cm2 V−1 s−1 in TGBC OTFT devices without CsF modification. Subsequently, the same group developed an asymmetry BTI derivative, BTzTI, by replacing one thiophene unit in the BTI core with a thiazole moiety to address the relatively high-lying HOMO energy level of BTI-based A–A type polymers and tackle the problem of low ordered thin-films for P87.163 In addition, to ensure the regioregularity of the polymer backbone and overcome the low reactivity of the C–Br bond on the thiazole unit, the BTzTI unit was first dimerized and the resulting dimer (DTzTI) was polymerized by using Stille polycondensation. As a result, the A–A type polymer P88 with a decent Mn of 7.0 kDa was obtained.163 The polymer showed a low-lying HOMO energy level of −5.78 eV, and highly ordered thin-films as revealed by the presence of diffraction peaks up to the fifth order in film XRD patterns. Consequently, P88 exhibited a remarkably high μe of 1.61 cm2 V−1 s−1 when TGBC device configuration was employed.
Based on the amide containing isoindigo (IID) and thienopyrroledione (TPD), P89 and P90 were synthesized via DArP by Leclerc et al.156 This was the first report on A–A type polymers made by DArP. After detailed optimization of the reaction conditions including the ligand and base, P89 and P90 with rather high Mns of 20 and 24 kDa, respectively, were obtained. However, using standard Suzuki polycondensation produced no polymeric product for P89 and low Mn product for P90 (13 kDa), implying the advantages of DArP for the synthesis of A–A type polymers. BGBC OTFT devices with P89 and P90 as semiconductor materials showed unipolar electron transport behaviour, and the maximum μe for P89 and P90 were 3.5 × 10−3 and 3.0 × 10−4 cm2 V−1 s−1, respectively. Subsequently, A–A type polymers P90-1 and P91 comprising IID-TPD and IID-BT, respectively, were synthesized by Yang et al., using Suzuki polycondensation.153 Both polymers showed similar HOMO energy levels of ∼−5.70 eV, while the LUMO energy level of P90-1 (−3.66 eV) was obviously lower than that of P91 (−3.54 eV). This indicates that the TPD unit has a stronger electron-withdrawing ability compared to the BT unit. After annealing at 150 °C, P91 exhibited a μe of 0.22 cm2 V−1 s−1. On the other hand, about one order of magnitude lower μe was obtained for the annealed P90-1 film (∼0.01 cm2 V−1 s−1). The poorer device performance of P90-1 is presumably attributed to the smaller grains and lower degree of molecular packing in thin-films. Embedding I–N in the IID unit could significantly improve the planarity of the polymer backbone and hence enhance the molecular packing order in thin-films. Thus, McCulloch et al. synthesized P92 via coupling diazaisoindigo (AIID) with a BT unit.154 Thanks to the more ordered thin-film and lower energetic disorder enabled by the highly planar backbones, high μe of up to 1.0 cm2 V−1 s−1 was achieved for the devices based on P92.
Ober et al. reported P93 comprising benzotriazole and BT units,145 which was synthesized by Suzuki polycondensation between dipotassium bis(trifluoroborate) of 2-alkylbenzotriazole and dibromobenzothiadiazole. By using LiOH as base, which can effectively hydrolyze trifluoroborate to the boronic acid, P93 with a high Mn of up to 120 kDa was obtained. P93 showed a unipolar electron transport characteristic with a μe of 0.020 cm2 V−1 s−1.
Swager et al. synthesized an A–A type polymer P94 carrying perfluorinated side-chains via Stille polycondensation.151 P94 was only soluble in perfluorinated solvents and insoluble in common organic solvents. This feature would allow for orthogonal processing of devices with multi-layered configurations. P94-based TGBC OTFT devices showed a μe of 2.2 × 10−6 cm2 V−1 s−1.
Conclusion and outlook
In this review, we have summarized the recent progress of n-type CPs from the aspect of molecule design. It is clear that controlling the HOMO/LUMO energy levels and orbital distributions of CPs are critical to achieve unipolar n-type transport behaviour. In view of these key points, four molecule design strategies were outlined and discussed in detail, i.e., D–A CPs with strong A units, D–A CPs with weak D units, D–A CPs with dual-A and A–A type CPs. On the basis of energetic consideration, the strategies of “D–A CPs with weak D units” and “A–A type CPs” are highly desired for constructing n-type CPs because they can eliminate the adverse effect of an electron-rich D unit, which tends to elevate the HOMO energy levels of the polymers and thus reduce the hole injection barriers. Additionally, although we classified some polymers into one specific strategy, several polymers were designed by multiple strategies. For instance, P40 is synthesized by the combination of “dual-A” and “weak D” design strategies. In fact, multiple strategies may be more effective than a single strategy in terms of energy level modulation. We hope that the findings summarized in this review will provide some guidelines for further development of high-performance n-type CPs and accelerate their applications in organic electronics.Although some of the n-type CPs have exhibited comparable performance with their p-type counterparts, the overall development of n-type CPs still lags far behind p-type CPs. Improving the electron mobility is a common theme in the development of n-type CPs because the range of possible applications for OTFT devices increases with the enhancement of mobility. Besides, following challenges also deserve further attention: (1) there are still very few purely unipolar n-type CPs. Many reported n-type CPs also exhibited weak hole transport characteristics as evidenced by the lack of clear off-regime in their transfer curves and the observation of linear increased current in their output curves at the low gate voltage and high source–drain voltage. This undesired hole transport will result in difficult switch off and high power consumption of the devices.21,105 (2) Most of the high-performance n-type CPs reported so far are only dissolved in chlorinated solvents, such as chloroform and dichlorobenzene, due to their strong intermolecular interactions. Processing with “greener” non-chlorinated solvents is necessary for the practical use of polymeric semiconductors in printing electronics. Therefore, the development of polymeric semiconductors that can be processed with non-chlorinated solvents is still a critical issue. (3) Uniaxial alignment of conjugated polymer backbones in thin-films can significantly enhance the charge carrier mobility of OTFT devices. Several techniques, such as dip coating, off-centre spin coating, shear coating and wire-bar coating, have been reported for the preparation of uniaxially aligned films of CPs. Among them, shear coating and wire-bar coating are highly desirable because they can be adopted in large-scale manufacturing processes. It was found that the alignment of polymer backbones produced by these deposition techniques was closely related to the aggregation of polymer chains in solution.164 Therefore, rational design of CPs to control the aggregation structures in solution is necessary. In addition, new professional techniques to characterize solution-state supramolecular structures should be developed. In this regard, a recent work reported by Pei et al. provided a guideline.81 (4) Mass production of high-performance CPs with small batch-to-batch variation is still a great challenge. Most of the reported high mobility CPs are synthesized via traditional polycondensation, such as Stille polycondensation, which requires the preparation of an organotin monomer by using a flammable organolithium reagent and produces a stoichiometric amount of toxic by-products. Furthermore, as we discussed above, polymers with an A–A backbone are highly desirable for the development of n-type CPs. However, A–A type polymers with high Mn, which frequently provide more efficient electron transport, usually cannot be obtained by the traditional polycondensation methods. Therefore, an efficient and environmentally benign synthetic strategy must be developed. DArP is emerging as an alternative protocol to traditional polycondensation methods for the synthesis of CPs.157,165–167 However, DArP is greatly restricted by the limited substrate scope and the narrow applicability of reaction conditions.157,165–167 It is obvious that there are significant opportunities to develop high-performance n-type CPs by combining synthetic chemistry with molecule design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21774093) and the National Key R&D Program of “Strategic Advanced Electronic Materials” (No. 2016YFB0401100) of the Chinese Ministry of Science and Technology.Notes and references
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- K. Fukuda, Y. Takeda, Y. Yoshimura, R. Shiwaku, L. T. Tran, T. Sekine, M. Mizukami, D. Kumaki and S. Tokito, Nat. Commun., 2014, 5, 4147 CrossRef CAS PubMed.
- X. Gao and Z. Zhao, Sci. China: Chem., 2015, 58, 947–968 CrossRef CAS.
- L. Shi, Y. Guo, W. Hu and Y. Liu, Mater. Chem. Front., 2017, 1, 2423–2456 RSC.
- I. Osaka and K. Takimiya, Adv. Mater., 2017, 29, 1605218 CrossRef PubMed.
- Z. Liu, G. Zhang and D. Zhang, Acc. Chem. Res., 2018, 51, 1422–1432 CrossRef CAS PubMed.
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748–2785 CAS.
- M. Muccini, Nat. Mater., 2006, 5, 605 CrossRef CAS PubMed.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2018, 6, 10390–10410 RSC.
- J. Xu, S. Wang, G.-J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y.-H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B.-H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59–64 CrossRef CAS PubMed.
- C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J.-L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728–3743 RSC.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099–3117 RSC.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS PubMed.
- J. Choi, H. Song, N. Kim and F. S. Kim, Semicond. Sci. Technol., 2015, 30, 064002 CrossRef.
- J. T. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- R. J. Baker, CMOS Circuit Design, Layout, and Simulation, John Wiley & Sons, 2010, pp. 6–7 Search PubMed.
- K. J. Baeg, M. Caironi and Y. Y. Noh, Adv. Mater., 2013, 25, 4210–4244 CrossRef CAS PubMed.
- B. Sun, W. Hong, C. Guo, S. Sutty, H. Aziz and Y. Li, Org. Electron., 2016, 37, 190–196 CrossRef CAS.
- D. De Leeuw, M. Simenon, A. Brown and R. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS.
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2017, 29, 1606217 CrossRef PubMed.
- Y. Sui, Y. Deng, Y. Han, J. Zhang, W. Hu and Y. Geng, J. Mater. Chem. C, 2018, 6, 12896–12903 RSC.
- L. Zhang, Z. Wang, C. Duan, Z. Wang, Y. Deng, J. Xu, F. Huang and Y. Cao, Chem. Mater., 2018, 30, 8343–8351 CrossRef CAS.
- F. Chen, Y. Jiang, Y. Sui, J. Zhang, H. Tian, Y. Han, Y. Deng, W. Hu and Y. Geng, Macromolecules, 2018, 51, 8652–8661 CrossRef CAS.
- H. T. Nicolai, M. Kuik, G. Wetzelaer, B. De Boer, C. Campbell, C. Risko, J. Brédas and P. Blom, Nat. Mater., 2012, 11, 882 CrossRef CAS PubMed.
- G. W. van Pruissen, E. A. Pidko, M. M. Wienk and R. A. Janssen, J. Mater. Chem. C, 2014, 2, 731–735 RSC.
- K. Kawabata, I. Osaka, M. Nakano, N. Takemura, T. Koganezawa and K. Takimiya, Adv. Electron. Mater., 2015, 1, 1500039 CrossRef.
- K. Kawabata, M. Saito, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 7725–7732 CrossRef CAS PubMed.
- H. Song, Y. Deng, Y. Gao, Y. Jiang, H. Tian, D. Yan, Y. Geng and F. Wang, Macromolecules, 2017, 50, 2344–2353 CrossRef CAS.
- X. Lv, W. Li, M. Ouyang, Y. Zhang, D. S. Wright and C. Zhang, J. Mater. Chem. C, 2017, 5, 12–28 RSC.
- Y.-W. Su, Y.-C. Lin and K.-H. Wei, J. Mater. Chem. A, 2017, 5, 24051–24075 RSC.
- Z. Yuan, B. Fu, S. Thomas, S. Zhang, G. DeLuca, R. Chang, L. Lopez, C. Fares, G. Zhang, J.-L. Bredas and E. Reichmanis, Chem. Mater., 2016, 28, 6045–6049 CrossRef CAS.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- R. S. Ashraf, A. J. Kronemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- G. K. Dutta, A. R. Han, J. Lee, Y. Kim, J. H. Oh and C. Yang, Adv. Funct. Mater., 2013, 23, 5317–5325 CrossRef CAS.
- G. Kim, A.-R. Han, H. R. Lee, J. Lee, J. H. Oh and C. Yang, Chem. Commun., 2014, 50, 2180–2183 RSC.
- A. Babel and S. A. Jenekhe, J. Am. Chem. Soc., 2003, 125, 13656–13657 CrossRef CAS PubMed.
- Y. Wang, T. Hasegawa, H. Matsumoto and T. Michinobu, J. Am. Chem. Soc., 2019, 141, 3566–3575 CrossRef CAS PubMed.
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–687 CrossRef CAS PubMed.
- H. Opoku, B. Nketia-Yawson, E. S. Shin and Y.-Y. Noh, Org. Electron., 2017, 41, 198–204 CrossRef CAS.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- Y. Kim, D. X. Long, J. Lee, G. Kim, T. J. Shin, K.-W. Nam, Y.-Y. Noh and C. Yang, Macromolecules, 2015, 48, 5179–5187 CrossRef CAS.
- B. Kang, R. Kim, S. B. Lee, S.-K. Kwon, Y.-H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef CAS PubMed.
- F. So, World Scientific Handbook of Organic Optoelectronic Devices (Volumes 1 & 2): Volume 1: Perovskite Electronics Volume 2: Organic Photovoltaics (OPVs), World Scientific, 2018 Search PubMed.
- Y.-J. Hwang, G. Ren, N. M. Murari and S. A. Jenekhe, Macromolecules, 2012, 45, 9056–9062 CrossRef CAS.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- I. Kang, T. K. An, J. A. Hong, H. J. Yun, R. Kim, D. S. Chung, C. E. Park, Y. H. Kim and S. K. Kwon, Adv. Mater., 2013, 25, 524–528 CrossRef CAS PubMed.
- R. Kim, P. S. K. Amegadze, I. Kang, H. J. Yun, Y. Y. Noh, S. K. Kwon and Y. H. Kim, Adv. Funct. Mater., 2013, 23, 5719–5727 CrossRef CAS.
- M. J. Sung, A. Luzio, W. T. Park, R. Kim, E. Gann, F. Maddalena, G. Pace, Y. Xu, D. Natali, C. de Falco, L. Dang, C. R. McNeill, M. Caironi, Y. Y. Noh and Y. H. Kim, Adv. Funct. Mater., 2016, 26, 4984–4997 CrossRef CAS.
- H. Chen, Y. Guo, Z. Mao, G. Yu, J. Huang, Y. Zhao and Y. Liu, Chem. Mater., 2013, 25, 3589–3596 CrossRef CAS.
- D. Fazzi, M. Caironi and C. Castiglioni, J. Am. Chem. Soc., 2011, 133, 19056–19059 CrossRef CAS PubMed.
- T. Schuettfort, S. Huettner, S. Lilliu, J. E. Macdonald, L. Thomsen and C. R. McNeill, Macromolecules, 2011, 44, 1530–1539 CrossRef CAS.
- R. Steyrleuthner, M. Schubert, I. Howard, B. Klaumünzer, K. Schilling, Z. Chen, P. Saalfrank, F. Laquai, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2012, 134, 18303–18317 CrossRef CAS PubMed.
- Z. Fei, Y. Han, J. Martin, F. H. Scholes, M. Al-Hashimi, S. Y. AlQaradawi, N. Stingelin, T. D. Anthopoulos and M. Heeney, Macromolecules, 2016, 49, 6384–6393 CrossRef CAS.
- T. Sajoto, S. P. Tiwari, H. Li, C. Risko, S. Barlow, Q. Zhang, J.-Y. Cho, J.-L. Brédas, B. Kippelen and S. R. Marder, Polymer, 2012, 53, 1072–1078 CrossRef CAS.
- M. He, W. Li, Y. Gao, H. Tian, J. Zhang, H. Tong, D. Yan, Y. Geng and F. Wang, Macromolecules, 2016, 49, 825–832 CrossRef CAS.
- Y. Deng, Y. Chen, X. Zhang, H. Tian, C. Bao, D. Yan, Y. Geng and F. Wang, Macromolecules, 2012, 45, 8621–8627 CrossRef CAS.
- C.-E. Tsai, R.-H. Yu, F.-J. Lin, Y.-Y. Lai, J.-Y. Hsu, S.-W. Cheng, C.-S. Hsu and Y.-J. Cheng, Chem. Mater., 2016, 28, 5121–5130 CrossRef CAS.
- P. Prins, F. Grozema, J. Schins, S. Patil, U. Scherf and L. Siebbeles, Phys. Rev. Lett., 2006, 96, 146601 CrossRef CAS PubMed.
- M. Yuan, M. M. Durban, P. D. Kazarinoff, D. F. Zeigler, A. H. Rice, Y. Segawa and C. K. Luscombe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4061–4069 CrossRef CAS.
- Z. Zhao, F. Zhang, Y. Hu, Z. Wang, B. Leng, X. Gao, C.-A. Di and D. Zhu, ACS Macro Lett., 2014, 3, 1174–1177 CrossRef CAS.
- X. Zhan, Z. a. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- L. Zhang, C. a. Di, Y. Zhao, Y. Guo, X. Sun, Y. Wen, W. Zhou, X. Zhan, G. Yu and Y. Liu, Adv. Mater., 2010, 22, 3537–3541 CrossRef CAS PubMed.
- X. Zhao, L. Ma, L. Zhang, Y. Wen, J. Chen, Z. Shuai, Y. Liu and X. Zhan, Macromolecules, 2013, 46, 2152–2158 CrossRef CAS.
- W. Zhou, Y. Wen, L. Ma, Y. Liu and X. Zhan, Macromolecules, 2012, 45, 4115–4121 CrossRef CAS.
- S. K. Samanta, I. Song, J. H. Yoo and J. H. Oh, ACS Appl. Mater. Interfaces, 2018, 10, 32444–32453 CrossRef CAS PubMed.
- Z. Yan, B. Sun and Y. Li, Chem. Commun., 2013, 49, 3790–3792 RSC.
- T. Lei, J.-H. Dou, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2013, 135, 12168–12171 CrossRef CAS PubMed.
- Y. Wang, S. D. Motta, F. Negri and R. Friedlein, J. Am. Chem. Soc., 2011, 133, 10054–10057 CrossRef CAS PubMed.
- J. H. Dou, Y. Q. Zheng, T. Lei, S. D. Zhang, Z. Wang, W. B. Zhang, J. Y. Wang and J. Pei, Adv. Funct. Mater., 2014, 24, 6270–6278 CrossRef CAS.
- T. Lei, J. H. Dou, X. Y. Cao, J. Y. Wang and J. Pei, Adv. Mater., 2013, 25, 6589–6593 CrossRef CAS PubMed.
- Y. Q. Zheng, Z. F. Yao, T. Lei, J. H. Dou, C. Y. Yang, L. Zou, X. Meng, W. Ma, J. Y. Wang and J. Pei, Adv. Mater., 2017, 29, 1701072 CrossRef PubMed.
- G. Zhang, J. Guo, M. Zhu, P. Li, H. Lu, K. Cho and L. Qiu, Polym. Chem., 2015, 6, 2531–2540 RSC.
- X. Zhou, N. Ai, Z.-H. Guo, F.-D. Zhuang, Y.-S. Jiang, J.-Y. Wang and J. Pei, Chem. Mater., 2015, 27, 1815–1820 CrossRef CAS.
- T. Lei, X. Xia, J.-Y. Wang, C.-J. Liu and J. Pei, J. Am. Chem. Soc., 2014, 136, 2135–2141 CrossRef CAS PubMed.
- Y. Q. Zheng, T. Lei, J. H. Dou, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, Adv. Mater., 2016, 28, 7213–7219 CrossRef CAS PubMed.
- Y.-Z. Dai, N. Ai, Y. Lu, Y.-Q. Zheng, J.-H. Dou, K. Shi, T. Lei, J.-Y. Wang and J. Pei, Chem. Sci., 2016, 7, 5753–5757 RSC.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636–2642 CrossRef CAS PubMed.
- Y. Kim, H. Hwang, N. K. Kim, K. Hwang, J. J. Park, G. I. Shin and D. Y. Kim, Adv. Mater., 2018, 30, 1706557 CrossRef PubMed.
- W. Hong, C. Guo, Y. Li, Y. Zheng, C. Huang, S. Lu and A. Facchetti, J. Mater. Chem., 2012, 22, 22282–22289 RSC.
- J. W. Rumer, M. Levick, S.-Y. Dai, S. Rossbauer, Z. Huang, L. Biniek, T. D. Anthopoulos, J. R. Durrant, D. J. Procter and I. McCulloch, Chem. Commun., 2013, 49, 4465–4467 RSC.
- W. Cui and F. Wudl, Macromolecules, 2013, 46, 7232–7238 CrossRef CAS.
- H. Hwang, D. Khim, J. M. Yun, E. Jung, S. Y. Jang, Y. H. Jang, Y. Y. Noh and D. Y. Kim, Adv. Funct. Mater., 2015, 25, 1146–1156 CrossRef CAS.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 3459–3462 CrossRef CAS PubMed.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 CrossRef PubMed.
- Y. Deng, J. Quinn, B. Sun, Y. He, J. Ellard and Y. Li, RSC Adv., 2016, 6, 34849–34854 RSC.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753–6759 CrossRef CAS PubMed.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- C. J. Mueller, C. R. Singh, M. Fried, S. Huettner and M. Thelakkat, Adv. Funct. Mater., 2015, 25, 2725–2736 CrossRef CAS.
- H.-Y. Chen, M. Nikolka, A. Wadsworth, W. Yue, A. Onwubiko, M. Xiao, A. J. P. White, D. Baran, H. Sirringhaus and I. McCulloch, Macromolecules, 2017, 51, 71–79 CrossRef.
- Y. Wang, Z. Yan, H. Guo, M. A. Uddin, S. Ling, X. Zhou, H. Su, J. Dai, H. Y. Woo and X. Guo, Angew. Chem., Int. Ed., 2017, 56, 15304–15308 CrossRef CAS PubMed.
- Y. Shi, H. Guo, M. Qin, Y. Wang, J. Zhao, H. Sun, H. Wang, Y. Wang, X. Zhou, A. Facchetti, X. Lu, M. Zhou and X. Guo, Chem. Mater., 2018, 30, 7988–8001 CrossRef CAS.
- Y. Ie, M. Nitani, M. Karakawa, H. Tada and Y. Aso, Adv. Funct. Mater., 2010, 20, 907–913 CrossRef CAS.
- I. Osaka, R. Zhang, J. Liu, D.-M. Smilgies, T. Kowalewski and R. D. McCullough, Chem. Mater., 2010, 22, 4191–4196 CrossRef CAS.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- K. Guo, J. Bai, Y. Jiang, Z. Wang, Y. Sui, Y. Deng, Y. Han, H. Tian and Y. Geng, Adv. Funct. Mater., 2018, 28, 1801097 CrossRef.
- J. Chen, X. Zhang, G. Wang, M. A. Uddin, Y. Tang, Y. Wang, Q. Liao, A. Facchetti, T. J. Marks and X. Guo, J. Mater. Chem. C, 2017, 5, 9559–9569 RSC.
- F. P. Wu, H. I. Un, Y. Li, H. Hu, Y. Yuan, B. Yang, K. Xiao, W. Chen, J. Y. Wang, Z. Q. Jiang, J. Pei and L. S. Liao, Chem. – Eur. J., 2017, 23, 14723–14727 CrossRef CAS PubMed.
- F. Wang, Y. Dai, W. Wang, H. Lu, L. Qiu, Y. Ding and G. Zhang, Chem. Mater., 2018, 30, 5451–5459 CrossRef CAS.
- A. Luzio, D. Fazzi, F. Nübling, R. Matsidik, A. Straub, H. Komber, E. Giussani, S. E. Watkins, M. Barbatti, W. Thiel, E. Gann, L. Thomsen, C. R. McNeill, M. Caironi and M. Sommer, Chem. Mater., 2014, 26, 6233–6240 CrossRef CAS.
- K. Kranthiraja, D. X. Long, V. G. Sree, W. Cho, Y.-R. Cho, A. Zaheer, J.-C. Lee, Y.-Y. Noh and S.-H. Jin, Macromolecules, 2018, 51, 5530–5536 CrossRef CAS.
- H. Xin, C. Ge, X. Jiao, X. Yang, K. Rundel, C. R. McNeill and X. Gao, Angew. Chem., Int. Ed., 2018, 57, 1322–1326 CrossRef CAS PubMed.
- Y. Wang, A. T.-R. Tan, T. Mori and T. Michinobu, J. Mater. Chem. C, 2018, 6, 3593–3603 RSC.
- H. S. Kim, G. Huseynova, Y.-Y. Noh and D.-H. Hwang, Macromolecules, 2017, 50, 7550–7558 CrossRef CAS.
- H. Usta, W. C. Sheets, M. Denti, G. Generali, R. Capelli, S. Lu, X. Yu, M. Muccini and A. Facchetti, Chem. Mater., 2014, 26, 6542–6556 CrossRef CAS.
- B. Fu, C.-Y. Wang, B. D. Rose, Y. Jiang, M. Chang, P.-H. Chu, Z. Yuan, C. Fuentes-Hernandez, B. Kippelen, J.-L. Bredas, D. M. Collard and E. Reichmanis, Chem. Mater., 2015, 27, 2928–2937 CrossRef CAS.
- C. Guo, J. Quinn, B. Sun and Y. Li, Polym. Chem., 2016, 7, 4515–4524 RSC.
- Y. Li, P. Sonar, S. P. Singh, M. S. Soh, M. van Meurs and J. Tan, J. Am. Chem. Soc., 2011, 133, 2198–2204 CrossRef CAS PubMed.
- H. N. Hong, H. J. Kim, A. Kim, S. Choi, Y. U. Kim, M. J. Cho and D. H. Choi, Synth. Met., 2018, 236, 1–7 CrossRef CAS.
- S. Shi, Y. Wang, M. A. Uddin, X. Zhou, H. Guo, Q. Liao, X. Zhu, X. Cheng, H. Y. Woo and X. Guo, Adv. Electron. Mater., 2017, 3, 1700100 CrossRef.
- S. Ma, G. Zhang, F. Wang, Y. Dai, H. Lu, L. Qiu, Y. Ding and K. Cho, Macromolecules, 2018, 51, 5704–5712 CrossRef CAS.
- Z. Yuan, C. Buckley, S. Thomas, G. Zhang, I. Bargigia, G. Wang, B. Fu, C. Silva, J.-L. Brédas and E. Reichmanis, Macromolecules, 2018, 51, 7320–7328 CrossRef CAS.
- R. Matsidik, M. Giorgio, A. Luzio, M. Caironi, H. Komber and M. Sommer, Eur. J. Org. Chem., 2018, 6121–6126 CrossRef CAS.
- M. Saito, I. Osaka, Y. Suda, H. Yoshida and K. Takimiya, Adv. Mater., 2016, 28, 6921–6925 CrossRef CAS PubMed.
- C. Zhu, Z. Zhao, H. Chen, L. Zheng, X. Li, J. Chen, Y. Sun, F. Liu, Y. Guo and Y. Liu, J. Am. Chem. Soc., 2017, 139, 17735–17738 CrossRef CAS PubMed.
- J. Yang, H. Wang, J. Chen, J. Huang, Y. Jiang, J. Zhang, L. Shi, Y. Sun, Z. Wei, G. Yu, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 1606162 CrossRef PubMed.
- L. Xu, Z. Zhao, M. Xiao, J. Yang, J. Xiao, Z. Yi, S. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 40549–40555 CrossRef CAS PubMed.
- J. Rumer, S. Rossbauer, M. Planells, S. Watkins, T. Anthopoulos and I. McCulloch, J. Mater. Chem. C, 2014, 2, 8822–8828 RSC.
- Z. A. Wang, J. Kuwabara, A. Ichige, T. Yasuda and T. Kanbara, Synth. Met., 2016, 222, 383–387 CrossRef CAS.
- Z. Zhao, Z. Yin, H. Chen, L. Zheng, C. Zhu, L. Zhang, S. Tan, H. Wang, Y. Guo, Q. Tang and Y. Liu, Adv. Mater., 2017, 29, 1602410 CrossRef PubMed.
- B. R. Conrad, C. K. Chan, M. A. Loth, S. R. Parkin, X. Zhang, D. M. Delongchamp, J. E. Anthony and D. J. Gundlach, Appl. Phys. Lett., 2010, 97, 133306 CrossRef.
- K. Kawashima, I. Osaka and K. Takimiya, Chem. Mater., 2015, 27, 6558–6570 CrossRef CAS.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed.
- Y. Wang, T. Hasegawa, H. Matsumoto, T. Mori and T. Michinobu, Adv. Funct. Mater., 2017, 27, 1701486 CrossRef.
- Y. Wang, T. Hasegawa, H. Matsumoto, T. Mori and T. Michinobu, Adv. Mater., 2018, 30, 1707164 CrossRef PubMed.
- D. M. Hodgson, J. Witherington, B. A. Moloney, I. C. Richards and J.-L. Brayer, Synlett, 1995, 32–34 CrossRef CAS.
- T. Lei, J. Y. Wang and J. Pei, Chem. Mater., 2013, 26, 594–603 CrossRef.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- S. Park, M. H. Lee, K. S. Ahn, H. H. Choi, J. Shin, J. Xu, J. Mei, K. Cho, Z. Bao, D. R. Lee, M. S. Kang and D. H. Kim, Adv. Funct. Mater., 2016, 26, 4627–4634 CrossRef CAS.
- G. Xue, X. Zhao, G. Qu, T. Xu, A. Gumyusenge, Z. Zhang, Y. Zhao, Y. Diao, H. Li and J. Mei, ACS Appl. Mater. Interfaces, 2017, 9, 25426–25433 CrossRef CAS PubMed.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS PubMed.
- S. Fabiano, H. Yoshida, Z. Chen, A. Facchetti and M. A. Loi, ACS Appl. Mater. Interfaces, 2013, 5, 4417–4422 CrossRef CAS PubMed.
- R. Steyrleuthner, M. Schubert, I. Howard, B. Klaumünzer, K. Schilling, Z. Chen, P. Saalfrank, F. Laquai, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2012, 134, 18303–18317 CrossRef CAS PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed.
- K. Guo, Y. Jiang, Y. Sui, Y. Deng and Y. Geng, Chin. J. Polym. Sci., 2019 DOI:10.1007/s10118-019-2277-1.
- J.-K. Lee, M. C. Gwinner, R. Berger, C. Newby, R. Zentel, R. H. Friend, H. Sirringhaus and C. K. Ober, J. Am. Chem. Soc., 2011, 133, 9949–9951 CrossRef CAS PubMed.
- M. M. Durban, P. D. Kazarinoff and C. K. Luscombe, Macromolecules, 2010, 43, 6348–6352 CrossRef CAS.
- J. A. Letizia, M. R. Salata, C. M. Tribout, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 9679–9694 CrossRef CAS PubMed.
- X. Guo, R. P. Ortiz, Y. Zheng, Y. Hu, Y. Y. Noh, K. J. Baeg, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 1405–1418 CrossRef CAS PubMed.
- Y. Wang, H. Guo, A. Harbuzaru, M. A. Uddin, I. Arrechea-Marcos, S. Ling, J. Yu, Y. Tang, H. Sun, J. T. López Navarrete, R. P. Ortiz, H. Y. Woo and X. Guo, J. Am. Chem. Soc., 2018, 140, 6095–6108 CrossRef CAS PubMed.
- Y. Shi, H. Guo, M. Qin, J. Zhao, Y. Wang, H. Wang, Y. Wang, A. Facchetti, X. Lu and X. Guo, Adv. Mater., 2018, 30, 1705745 CrossRef PubMed.
- Y. Takeda, T. L. Andrew, J. M. Lobez, A. J. Mork and T. M. Swager, Angew. Chem., Int. Ed., 2012, 51, 9042–9046 CrossRef CAS PubMed.
- S. Vasimalla, S. P. Senanayak, M. Sharma, K. S. Narayan and P. K. Iyer, Chem. Mater., 2014, 26, 4030–4037 CrossRef CAS.
- G. Kim, A.-R. Han, H. R. Lee, J. Lee, J. H. Oh and C. Yang, Chem. Commun., 2014, 50, 2180–2183 RSC.
- W. Yue, M. Nikolka, M. Xiao, A. Sadhanala, C. B. Nielsen, A. J. P. White, H. Y. Chen, A. Onwubiko, H. Sirringhaus and I. Mcculloch, J. Mater. Chem. C, 2016, 4, 9704–9710 RSC.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- F. Grenier, P. Berrouard, J. R. Pouliot, H. R. Tseng, A. J. Heeger and M. Leclerc, Polym. Chem., 2013, 4, 1836–1841 RSC.
- J. R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed.
- Y. Fukutomi, M. Nakano, J.-Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445–11448 CrossRef CAS PubMed.
- M. Nakano, I. Osaka and K. Takimiya, Macromolecules, 2015, 48, 576–584 CrossRef CAS.
- W. Yang, M. Nakano, T. Michinobu, Y. Kiyota, T. Mori and K. Takimiya, Macromolecules, 2017, 50, 857–864 CrossRef.
- X. Zhao, Y. Wen, L. Ren, L. Ma, Y. Liu and X. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4266–4271 CrossRef CAS.
- Y. Wang, H. Guo, S. Ling, I. Arrechea-Marcos, Y. Wang, J. T. López Navarrete, R. P. Ortiz and X. Guo, Angew. Chem., Int. Ed., 2017, 56, 9924–9929 CrossRef CAS PubMed.
- Y. Shi, H. Guo, M. Qin, J. Zhao, Y. Wang, H. Wang, Y. Wang, A. Facchetti, X. Lu and X. Guo, Adv. Mater., 2018, 30, 1705745 CrossRef PubMed.
- D. Khim, A. Luzio, G. E. Bonacchini, G. Pace, M. J. Lee, Y. Y. Noh and M. Caironi, Adv. Mater., 2018, 30, 1705463 CrossRef PubMed.
- T. Bura, J. T. Blaskovits and M. Leclerc, J. Am. Chem. Soc., 2016, 138, 10056 CrossRef CAS PubMed.
- H. Bohra and M. Wang, J. Mater. Chem. A, 2017, 5, 11550–11571 RSC.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2019 |