
A general low-temperature synthesis route to polyanionic vanadium phosphate fluoride cathode materials: AVPO4F (A = Li, Na, K) and Na3V2(PO4)2F3†
Nicolas
Goubard-Bretesché
a,
Erhard
Kemnitz
 a and
Nicola
Pinna
*ab
a and
Nicola
Pinna
*ab
aInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: nicola.pinna@hu-berlin.de
bIRIS Adlershof, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 21st August 2019
Abstract
Phosphate fluoride-based cathode materials attract great interest for secondary batteries as they offer a very rich crystal chemistry that allows one to finely tune the electrochemical properties. Among them, tavorite-LiVPO4F exhibits the highest theoretical energy density (655 W h kg−1) and is therefore strongly investigated in the field. However, the solid-state reactions reported for the synthesis of this material generally lead to a partial oxidation of the compound and the formation of side-products due to the evaporation of fluorine occurring at elevated temperature. Herein, we show that the use of a new liquid-phase fluorolytic route allows to tackle this issue and leads to phase-pure LiVPO4F at a low temperature of 240 °C. By slightly changing the synthesis parameters, we generalized this soft chemistry method to the related Na3V2(PO4)2F3 and (Na,K)VPO4F cathode materials that are also highly relevant for battery applications. The obtained materials exhibit good electrochemical performance, with almost full capacity reached at low rates. The versatility of the reported synthesis is therefore worth exploring for other polyanionic systems.
Introduction
Since the first report of the olivine-type LiFePO4 material by Goodenough and coworkers in 1997,1 three-dimensional frameworks based on the phosphate polyanion have attracted much attention in the battery community.2 As such, phosphate fluoride-based cathode materials are of great interest as they can combine the benefits of both their anionic species. The presence of highly covalent (PO4)3− groups provides them with a high structural stability and the inductive effect3,4 induced by both F− and (PO4)3− anions increases the potential of the transition metal ion redox couple. Different structural families of lithium, sodium and potassium transition metal phosphate–fluorides have been reported, such as AMPO4F,5,6 A2MPO4F,7 A3M2(PO4)2F3,8 and A5M(PO4)2F29 (in which A is an alkaline metal and M a transition metal), and their electrochemical performance has been investigated in the last two decades.Thanks to their very attractive working voltage (typically around 4 V in half cells) and decent theoretical capacity values (higher than 125 mA h g−1), vanadium-based phosphate fluorides (Li, Na, K)VPO4F10–12 and Na3V2(PO4)2F313,14 have been highly investigated in the literature. Among them, tavorite-type LiVPO4F displays a high V4+/V3+ redox potential (≈4.2 V vs. Li+/Li). Combined with a theoretical specific capacity of 156 mA h g−1, this leads to an energy density value of 655 W h kg−1vs. Li, which makes it very competitive with the already commercialized lithium iron phosphate (586 W h kg−1vs. Li for LiFePO4).15
However, synthesizing pure LiVPO4F is challenging. Typically, a carbothermal reduction (CTR) reaction is used,10 involving two successive heat treatments. First, a carbon powder is used as a reductive agent to obtain a VPO4/C composite material from a V5+ precursor (generally V2O5) at elevated temperature (typically ≥600 °C). Then, this product is reacted with LiF to obtain LiVPO4F/C, at high temperature, once again (≥600 °C in general). This second step is critical, as many parameters such as the amount of carbon in VPO4/C, the atmosphere, the heating time and the cooling rate must be precisely controlled to prevent the partial oxidation of the material in LiVPO4F1−xOx and the formation of impurities such as Li3V2(PO4)316,17 and V2O3.17 Other synthetic routes deriving from the CTR method have been reported using, for instance, oxalic acid18,19 or hydrogen20 as a reducing agent. Recently, Kim et al.21 reported for the first time a one-step synthesis of phase-pure LiVPO4F. To do so, they used polytetrafluoroethylene (PTFE) as an additional fluorine source to compensate its evaporation during the heat treatment, which is known to occur according to reaction (1):16,22
| 3LiVPO4F → Li3V2(PO4)3 + VF3(g) | (1) |
Another way to avoid the evaporation of volatile fluorine compounds during the synthesis of LiVPO4F is to use a “chimie douce” approach in a liquid medium, which can also offer the opportunity to better control the growth and morphology of the obtained particles, in comparison to solid-state routes. As such, the fluorolytic sol–gel route is a synthesis method that relies on the dissolution (or dispersion) of metal-ion precursors in a nonaqueous solvent (typically an alcohol), followed by the addition of hydrogen fluoride (HF) and heating of the solution. It can be generalized by the reaction (2):
| MXn + nHF → MFn + nHX | (2) |
Current efforts are made by our group toward the extension of this method to fluorine-containing battery-type materials.28–30 In this framework, we recently reported and discussed the fluorolytic sol–gel chemistry of polyanionic LiFePO4F and Li2CoPO4F materials.30 In the continuity of this work, we report herein a new straightforward one-pot solvothermal synthesis to phase-pure crystalline LiVPO4F at mild temperature (240 °C), thus suppressing the need for energy-consuming heat treatments at elevated temperatures under a controlled atmosphere. The obtained phase pure compound was characterized as a Li-ion cathode material and displays good electrochemical performance, reaching almost full theoretical capacity at low rate (147 mA h g−1 at C/10), without any carbon-coating at the surface of the particles that is usually achieved with the carbothermal route. While the emphasis of this paper is on the tavorite-LiVPO4F material, we also show that, by varying the synthesis conditions and the precursors, this new route can easily be generalized to the related sodiated and potassiated compounds NaVPO4F, Na3V2(PO4)2F3, and KVPO4F which are of great interest for sodium and potassium-ion batteries.11–13,31
Experimental
Reagents
Vanadium trichloride (VCl3, 99%), lithium methoxide (CH3OLi, >98%), sodium ethoxide (C2H5ONa, >96%) and potassium ethoxide (C2H5OK, >95%) were purchased from Alfa Aesar. Anhydrous benzyl alcohol (C6H5CH2OH, >99.8%) and crystalline phosphoric acid (H3PO4, 99.999%) were obtained from Sigma-Aldrich. All reagents were used without further purification.Synthesis of LiVPO4F material
The fluorolytic sol–gel synthesis route used to obtain LiVPO4F is adapted from our previous work on lithium iron/cobalt phosphate fluoride.30 Typically, vanadium trichloride, lithium methoxide and phosphoric acid (with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratio) were dissolved at 80 °C in benzyl alcohol to obtain a 0.2 M solution. Then, this latter was fluorinated by carefully adding a stoichiometric amount of a 20 M methanolic hydrofluoric acid (HF) solution, changing its color from dark violet to green. The mixture was subsequently stirred for a few minutes before being transferred into a Teflon-lined autoclave that was heated in a furnace at 240 °C for 12 h. To avoid the oxidation of V3+ during the synthesis, all steps were performed under argon by using a glovebox or Schlenk-line techniques. Increasing the duration of the heat treatment only induces a slight increase of the particle size.
:1:1 molar ratio) were dissolved at 80 °C in benzyl alcohol to obtain a 0.2 M solution. Then, this latter was fluorinated by carefully adding a stoichiometric amount of a 20 M methanolic hydrofluoric acid (HF) solution, changing its color from dark violet to green. The mixture was subsequently stirred for a few minutes before being transferred into a Teflon-lined autoclave that was heated in a furnace at 240 °C for 12 h. To avoid the oxidation of V3+ during the synthesis, all steps were performed under argon by using a glovebox or Schlenk-line techniques. Increasing the duration of the heat treatment only induces a slight increase of the particle size.
The obtained green powder was centrifuged and repeatedly resuspended in ultrapure water and ethanol. After a last resuspension in acetone, the material was dried at 65 °C for a few hours.
Similar results were obtained using vanadium acetylacetonate as a V(III) source.
Synthesis of KVPO4F material
KVPO4F was obtained using the same synthetic method as for LiVPO4F, but by replacing lithium methoxide by potassium ethoxide. The autoclave was heated at 240 °C for 24 h.Synthesis of Na3V2(PO4)2F3 and NaVPO4F materials
Both sodiated compounds were obtained using a similar route as for LiVPO4F. The lithium methoxide precursor was replaced by sodium ethoxide, and the ratios Na:V:P:F were adjusted to 3:2:2:3 and 1:1:1:1, for Na3V2(PO4)2F3 and NaVPO4F, respectively.
However, we found that in the case of NaVPO4F synthesis, the tetragonal Na3V2(PO4)2F3 phase was firstly crystallizing, prior to being progressively converted into the tavorite-type NaVPO4F, as already observed by Boivin et al.11 In this paper, the studied Na3V2(PO4)2F3 and NaVPO4F materials were obtained at 240 °C after 12 h and 72 h of solvothermal treatment, respectively.
Characterization
X-ray diffraction (XRD) patterns were acquired in transmission geometry using a STOE Stadi MP diffractometer equipped with a Dectris Mythen 1K linear silicon strip detector and a Ge(111) double-crystal monochromator with a Mo Kα1 radiation source (λ = 0.7093 Å).Le Bail refinements were made with the FullProf software.
Transmission Electron Microscopy (TEM) measurements were carried out using a FEI TALOS 200S equipped with a field emission gun and operated at 200 kV. A suspension of the desired material in absolute ethanol was prepared and a drop was deposited on a holey carbon-coated copper grid.
Energy dispersive X-ray spectroscopy (EDX) was recorded with build in SuperX EDS detector.
Fourier-transform infrared spectroscopy (FTIR) was carried out using a Thermo Scientific Nicolet iS5 FTIR Spectrometer in attenuated total reflection (ATR) configuration with a germanium crystal, in the range 4000–600 cm−1.
Electrochemical measurements
The working electrodes were made by mixing the desired active material with carbon black (Super C65, Timcal) in a 7:2 ratio at 400 rpm for 30 minutes using a planetary ball-mill (Pulverisette 7, Fritsch). The obtained black powder was then mixed with polyvinylidene fluoride (PVDF) in anhydrous N-methylpyrrolidone (NMP, Sigma-Aldrich, 99.5%) with a weight ratio of 9:1. The resulting slurry was subsequently casted onto an Al-foil using a doctor blade apparatus before being dried at 70 °C. After a cold-laminating step, disk-shaped electrodes of 18 mm diameter were cut and dried overnight at 120 °C under vacuum using a Büchi glass oven. The mass loading of all electrodes was comprised between 2 and 5 mg cm−2.
Coin cells were then assembled in an argon-filled glove box using the vanadium phosphate fluoride compound as a working electrode material and a lithium (or sodium) foil (Alfa Aesar, 99.9%) as both counter and reference electrodes. For lithium-based half-cells, a 1 M LiPF6 (ABCR, 99.9% battery grade) solution in a mix of ethylene carbonate (ABCR, 99.9%), propylene carbonate (ABCR, 99.9%,) and dimethyl carbonate (ABCR, 99.9%) with 1:1:1 volume ratio was used as the electrolyte, while a 1 M NaClO4 solution in PC was used for their sodium counterparts. In both cases, a glass fiber filter (Whatman) served as a separator.
Galvanostatic cycling was carried out at room temperature at different C-rates using a CT2001A battery cycler (Landt Instruments). A current density of 1C corresponds to the insertion/deinsertion of 1 alkali ion in 1 hour for (Li, Na, K)VPO4F, and of 2 Na for Na3V2(PO4)2F3 (theoretical capacity of 128 mA h g−1). The potential ranges used in this work were 2.5–4.6 V and 2.0–4.7 V vs. Li+/Li for LiVPO4F and KVPO4F, respectively. Na-ion half-cells were cycled from 2.5 to 4.5 V and 2.3 to 4.4 V vs. Na+/Na for NaVPO4F and Na3V2(PO4)2F3, respectively.
Results and discussion
LiVPO4F: fluorolytic sol–gel synthesis, structure and morphology
By adapting the synthesis conditions from our study of LiFePO4F,30 we successfully obtained phase-pure tavorite-LiVPO4F via a fluorolytic sol–gel route32,33 involving a solvothermal treatment for 12 h at 240 °C. As discussed in our previous work,30 H3PO4 is known to possess a strong complexing behavior towards metal cations.34 It tends therefore to quickly react with metal alkoxides and other metal precursors,35 to form M–O–P rather than M–O–M bonds. As such, during the synthesis of LiFePO4F, a poorly crystalline iron phosphate material was formed before the growth of the desired phosphate fluoride phase. Herein, we found that the lithium vanadium phosphate fluoride precursor solution was in fact a very stable sol and no precipitation could be observed, even after few days of storage and a thorough centrifugation step. After a solvothermal treatment of the sol, green aggregates of LiVPO4F powder embedded in a highly viscous translucent gel are obtained.The X-ray diffractogram (XRD) of the synthesized material and the schematic representation of its crystallographic structure are presented in Fig. 1a and b. The well-known tavorite framework, which is characteristic of many AxMPO4(OH, F) materials (in which A = Li, Na and M is typically a transition metal), consists in alternately tilted corner-sharing V3+O4F2 octahedra in the [101] direction, which share common fluorine atoms. These VO4F2 chains are linked to each other by corner-sharing PO4 tetrahedra, forming large (>3 Å wide) interconnecting channels along the [100] and [010] directions. Tavorite-type materials have been reported to be fast one-dimensional lithium ion conductors (along the [111] direction) by Mueller and colleagues,36 which makes them highly relevant as Li-ion battery cathode materials.
 | ||
Fig. 1 (a) Comparison of the experimental (open circles) and calculated (black line) XRD patterns for LiVPO4F, using a full-pattern matching (Le Bail) refinement in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The positions of the Bragg reflections (green markers) and the difference curve (blue line) are also shown. (b) Projections of the tavorite structure of LiVPO4F along the [100] and [010] directions. VO4F2 octahedra are represented in blue, PO4 tetrahedra in turquoise, and lithium atoms in yellow. . The positions of the Bragg reflections (green markers) and the difference curve (blue line) are also shown. (b) Projections of the tavorite structure of LiVPO4F along the [100] and [010] directions. VO4F2 octahedra are represented in blue, PO4 tetrahedra in turquoise, and lithium atoms in yellow. | ||
The XRD of the synthesized compound shows well-defined and intense peaks, indicating its high crystallinity. Le Bail refinements were performed in the P space group (tavorite-type structure) and the lattice parameters (a = 5.1699(1) Å, b = 5.3176(1) Å, c = 7.4729(1) Å, α = 112.645(1)°, β = 112.390(1)°, γ = 82.124(1)°) are similar to those reported by Ellis et al.37 An average crystallite size of 200 nm was calculated from the (1) and (11) planes (at 10.35° and 13.45°, respectively). No impurity peak is detectable in the diffractogram, whereas the generation of side-products such as Li3V2(PO4)3 or V2O3 is commonly reported for the solid-state synthesis of LiVPO4F, as discussed in the introduction section.
The transmission electron micrographs (Fig. 2a and b) show that the obtained material consists of agglomerates of well-faceted particles in the size range from few hundreds of nanometers to few micrometers. The HRTEM image (Fig. 2c) demonstrates the high crystallinity of the obtained material, which agrees well with the XRD results. Its power spectrum (Fig. 2d) can be attributed to the expected LiVPO4F phase oriented along the [00] zone axis. However, one should note that many reflections are possible for this structure and an orientation along the [10] zone axis is also a possibility. The atomic V:P:F ratio was determined by EDX and is close to the theoretical 1:1:1 value (1.0:1.04:0.98).
 | ||
| Fig. 2 TEM characterization of the obtained LiVPO4F material. (a and b) TEM images; (c) HRTEM image and (d) its corresponding power spectrum. | ||
The name “tavorite” originally comes from a natural mineral of chemical formula LiFePO4OH.38,39 This material is well known to be isostructural to its fluorinated counterpart,40 with only a slight variation in the lattice parameters. In a recent paper, Boivin et al.41 showed that it is also the case for the vanadium-related material, and successfully synthesized a LiVPO4OH tavorite compound via a hydrothermal route. Additionally, the related V4+-containing LiVPO4O42 and LiVPO4F1−xOx43,44 materials can also crystallize in the same crystalline structure. Therefore, to check the absence of any (OH)− group and of the oxidized phase in the obtained compound, a FTIR spectrum has been acquired and is shown in Fig. S1 (ESI†). The main absorption bands around 900–1200 cm−1 correspond to (PO4)3− groups.45 Interestingly, the O–H stretching signal (around 3000–3300 cm−1) is absent, which supports the expected fully fluorinated composition of LiVPO4F. This is one of the main advantages of using non-hydrolytic sol–gel chemistry: materials can be synthesized without intermediate hydroxyl groups thanks to aprotic condensation reactions.46 Additionally, vanadyl-type defects (V4+ = O), that should exhibit a stretching band around 875 cm−1 in the FTIR spectrum are not detected. Such a contribution of vanadyl defects has been detected by Boivin et al.43 even for a material they thought would be pure LiVPO4F. The fact that this feature is absent in our FTIR spectrum demonstrates the purity of our material and, therefore, also supports the LiVPO4F stoichiometry and V(III) oxidation state. We attribute such a behavior to the low-temperature non-hydrolytic synthetic route and the reducing properties of benzyl alcohol. Both could help preventing the oxidation of the vanadium47 that usually occurs in the material due to the heat treatments at high temperature that are employed during its synthesis.
Motivated by our previous success to synthesize the related tavorite LiFePO4F material in a few minutes by a microwave-assisted solvothermal route, we tried to apply this approach to LiVPO4F as well. To this end, we transferred 2.5 mL of the precursor solution in a 10 mL silicon carbide vial and applied microwave heating at 240 °C using an Anton Paar Monowave 300 equipment. We found that the crystallization of LiVPO4F is somehow much slower than its iron-containing counterpart. Indeed, after 40 min of microwave heating, a much less crystalline tavorite phase is formed, as demonstrated in the diffractogram in Fig. S2 (ESI†). However, one can note that no other crystalline phase was detected after such a short heating treatment, suggesting that LiVPO4F particles nucleate and grow directly, without forming any intermediate phase. To maximize the electrochemical performance, we chose the material with the highest crystallinity to manufacture the electrodes.
Electrochemical properties of LiVPO4F
Typical galvanostatic and differential capacity curves of LiVPO4F at C/10 rate are displayed in Fig. 3a and b. The charging process corresponds to the extraction of lithium from the structure of the compound, inducing an oxidation of V3+ to V4+ in the material. Two plateaus are visible on the electrochemical profiles, centered around 4.18 and 4.30 V vs. Li+/Li, respectively. On the other hand, only one plateau is visible at 4.21 V vs. Li+/Li during the discharge of the compound. This electrochemical profile is fully consistent with previous reports on LiVPO4F material10,17,21,37,43 and it is attributed to the formation of a Li0.67VPO4F intermediate phase, which only occurs during the delithiation step (LiVPO4F → Li0.67VPO4F → VPO4F), as demonstrated by Ateba Mba et al.17 This unusual asymmetrical electrochemical behavior is however not fully understood yet. In addition to these plateaus, a broad contribution can be seen between 3.7 and 4.0 V vs. Li+/Li. We attribute this feature to a partial amorphization of LiVPO4F, induced by the ball-milling step that was performed prior to manufacturing the electrodes, as we already demonstrated for tavorite LiFePO4F material synthesized using a similar route.30 In total, a high discharge capacity of 147 mA h g−1 is obtained, corresponding to the insertion of 0.94 Li+ per formula unit, (i.e. almost full capacity). The charge capacity (161 mA h g−1), however, exceeds the theoretical one, which is attributed to a partial oxidation of the electrolyte occurring at high potential.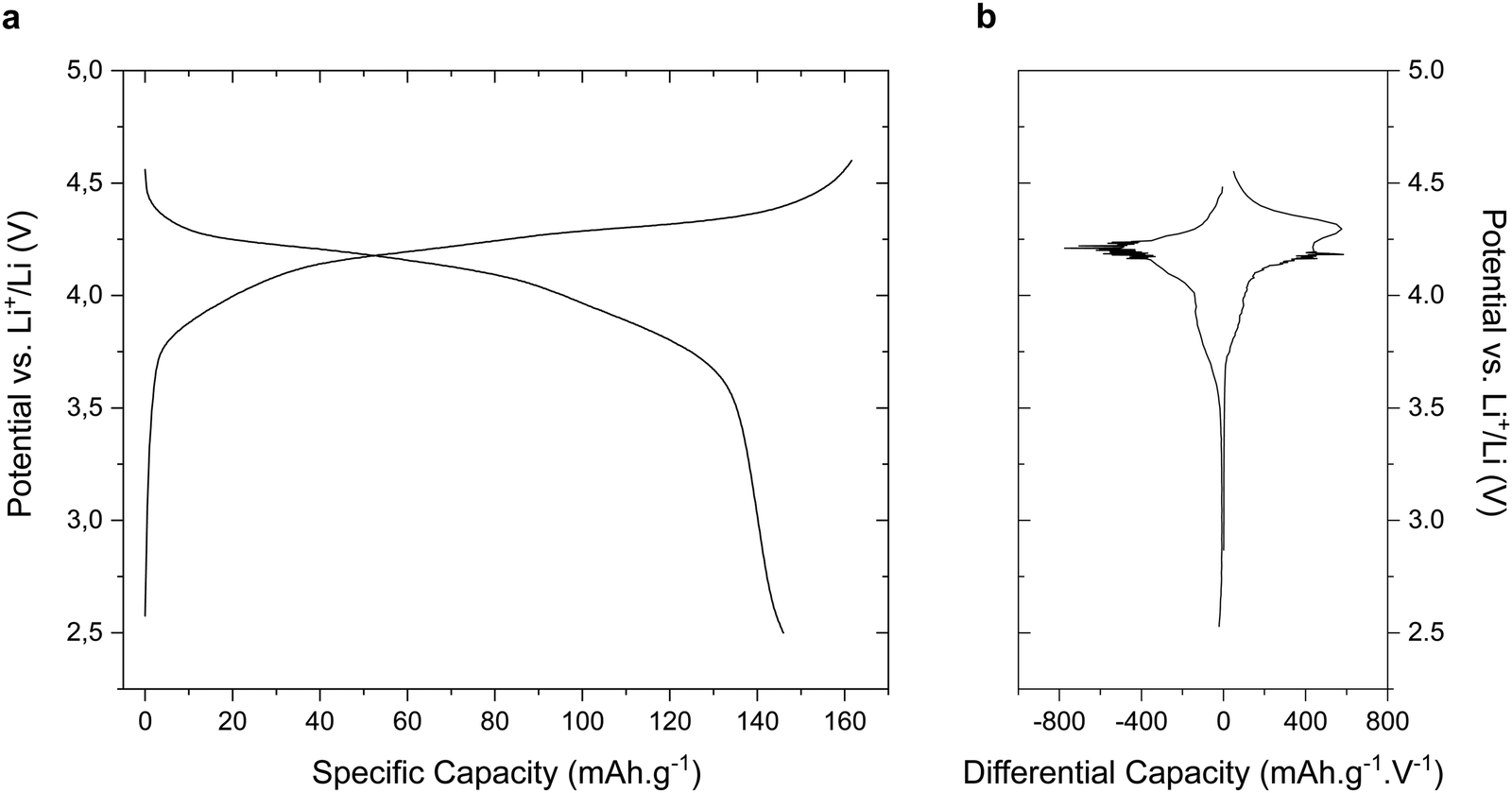 | ||
| Fig. 3 Typical galvanostatic cycling curve of LiVPO4F material synthesized via a fluorolytic sol–gel route (a) and its corresponding differential capacity profile (b). | ||
The galvanostatic curves and the corresponding capacity values of the LiVPO4F compound at various C-rates are depicted in Fig. 4a and b. The electrochemical behavior of the material at C/5 is very close to that at C/10 rate, with a specific capacity of 138 mA h g−1. However, its performance quickly decreases at higher rates, with capacity values of 102 mA h g−1 at C/2 and only 49 mA h g−1 retained at 1C rate. The low rate capability of the material can be attributed, at least partly, to the absence of carbon-coating on the LiVPO4F particles, which is inherent to our synthetic route. Nevertheless, low-temperature wet-chemistry routes, like the one reported herein, allow the introduction of carbon-based materials directly into the solution, which can lead to the formation of carbon-containing composite materials that could greatly enhance the high-rate electrochemical performance. This optimization part was, however, not the scope of this work but would certainly be beneficial to the rate capability of our LiVPO4F material. Additionally, one should note that such a behavior is not unusual and has even been reported for carbon-coated LiVPO4F electrodes.48 Indeed, previous studies reveal a high disparity in the rate capability obtained for this material. The reason for this is still under debate and some hypotheses involving the quality of the carbon-coating, the particle size and the vanadyl-type (V4+ = O) “defects” in the structure were proposed.21,48 Boivin et al.48 recently synthesized LiVPO4(F, O) materials with different fluorine/oxygen compositions, therefore inducing various amounts of vanadyl “defects” in the same crystallographic structure, and compared their electrochemical performance. They concluded that the introduction of a small amount of V4+ in the material is beneficial to its high rate behavior, whereas Kim et al.,21 on the contrary, attribute the high rate capability exhibited by their LiVPO4F material to its high purity.
 | ||
| Fig. 4 (a) Typical galvanostatic curves of LiVPO4F obtained at different C-rates; (b) corresponding evolution of the discharge capacities and coulombic efficiency of the material as a function of the cycle number. | ||
To get additional insight about the vanadyl-type defects content of our material, we decided to focus on the low-voltage parts of the galvanostatic curves, as proposed by Boivin et al.48 Indeed, both LiVPO4F and LiVPO4O materials are electrochemically active in the low-voltage range, but exhibit completely different behavior. The reduction of LiV3+PO4F in Li2V2+PO4F is known to occur at 1.8 V vs. Li+/Li,37 whereas the insertion of Li+ in LiVPO4O is accompanied by various phase transitions at 2.45, 2.21 and 2.04 V vs. Li+/Li.49 Studying the insertion of Li+ in our LiVPO4F material allows therefore to indirectly probe its purity. The galvanostatic and differential capacity curves of a LiVPO4F electrode in the low-voltage range are depicted in Fig. 5a and b. Unlike what could be expected for a pure LiVPO4F material, a small contribution centered around 2.5 V vs. Li+/Li can be observed, which is consistent with a partially oxidized LiVPO4F1−xOx material. Such a feature can also be seen when looking closely at the high-voltage region of the electrochemical curves (Fig. 4 and 5a), where a slight decrease of the slope is perceptible from ca. 2.7 V vs. Li+/Li during the Li insertion in the material, especially at low rate. The other phase transitions characteristic of Li+ insertion/deinsertion in LiVPO4O are known to generate less capacity (ca. two times less)17,49 than the first one, and can therefore only be guessed on the curves.
 | ||
| Fig. 5 Galvanostatic profiles and corresponding derivative curves of ball-milled (a and b) and pristine (c and d) LiVPO4F material, in the low-voltage range at C/10 rate. | ||
The unambiguous partial oxidation of LiVPO4F in our electrodes came as a surprise to us, as is was not consistent with the results obtained by using FTIR (no vanadyl bond observable) and EDX (ratio Li:V:P:F almost stoichiometric) spectroscopies. However, one should note that prior to making the electrodes, a ball-milling step was performed on a mixture of LiVPO4F and carbon black (with a 7:2 ratio, see the experimental section). Even though this important step was carried out in a recipient closed under argon atmosphere, one could expect a partial oxidation at the surface of the particles during or after this process. Consequently, electrodes were manufactured with the pristine LiVPO4F material using the same formulation, but without any ball-milling treatment. Galvanostatic cycling was performed in the low voltage region of the material and the obtained results (Fig. 5c and d) are consistent with this hypothesis. Only one plateau centered at 1.8 V vs. Li+/Li is visible on the electrochemical profile, which is characteristic of a pure LiVPO4F material. One can also note the difference in capacity and slope between the ball-milled and the pristine material. The balled-milled sample exhibits a much more sloping profile, which is typical for nanosized materials50 and consistent with our recent study of LiFePO4F.30 The low capacity of the pristine sample as compared to the ball-milled one can be assessed to the insulating character of LiVPO4F (electronic conductivity of ca. 10−11 S cm−1, according to Xiao et al.20) combined to a non-optimized contact between the conductive carbon and the LiVPO4F particles (absence of carbon coating and ball-milling treatment).
As stated above, the link between the physicochemical properties of LiVPO4F and its electrochemical performance is still under debate in the community, and the amount of V4+ in the material seems to be an important parameter. As no vanadyl-defect was detected in our pristine LiVPO4F material, we also evaluated its power performance in the high-voltage range (Fig. S3a and b, ESI†). Surprisingly, despite a larger particle size and a poor capacity at low rate, the power behavior of the pristine material is much improved compared to the ball-milled sample, with still 63% of the initial capacity sustained at 1C rate, compared to only 32% for the ball-milled material. Further experiments involving the introduction of a tuned amount of vanadyl defects in the structure while conserving the same morphology and synthetic route would however be necessary to definitely assess the reason for these different electrochemical behaviors.
Fluorolytic sol–gel synthesis extended to Na3V2(PO4)2F3, NaVPO4F, and KVPO4F related materials
The demonstration that LiVPO4F could be easily synthesized by soft-chemistry approaches encouraged us to try to extend our synthesis route to other related vanadium phosphate fluoride compounds. In this line, by slightly changing the synthesis parameters (i.e. alkaline metal precursor and reaction time, see the experimental section), we successfully obtained Na3V2(PO4)2F3, NaVPO4F and KVPO4F materials.Fig. 6 displays the X-ray diffractograms of the obtained powders, and the schematic polyhedral representations of their respective crystal structures. Similar to what was obtained for LiVPO4F, the patterns of all three samples are pure, single-phase and representative of well crystalline materials. By using Le Bail (full-pattern matching) refinements, we show that the X-ray diffraction peaks of all synthesized compounds could be indexed to their expected frameworks.
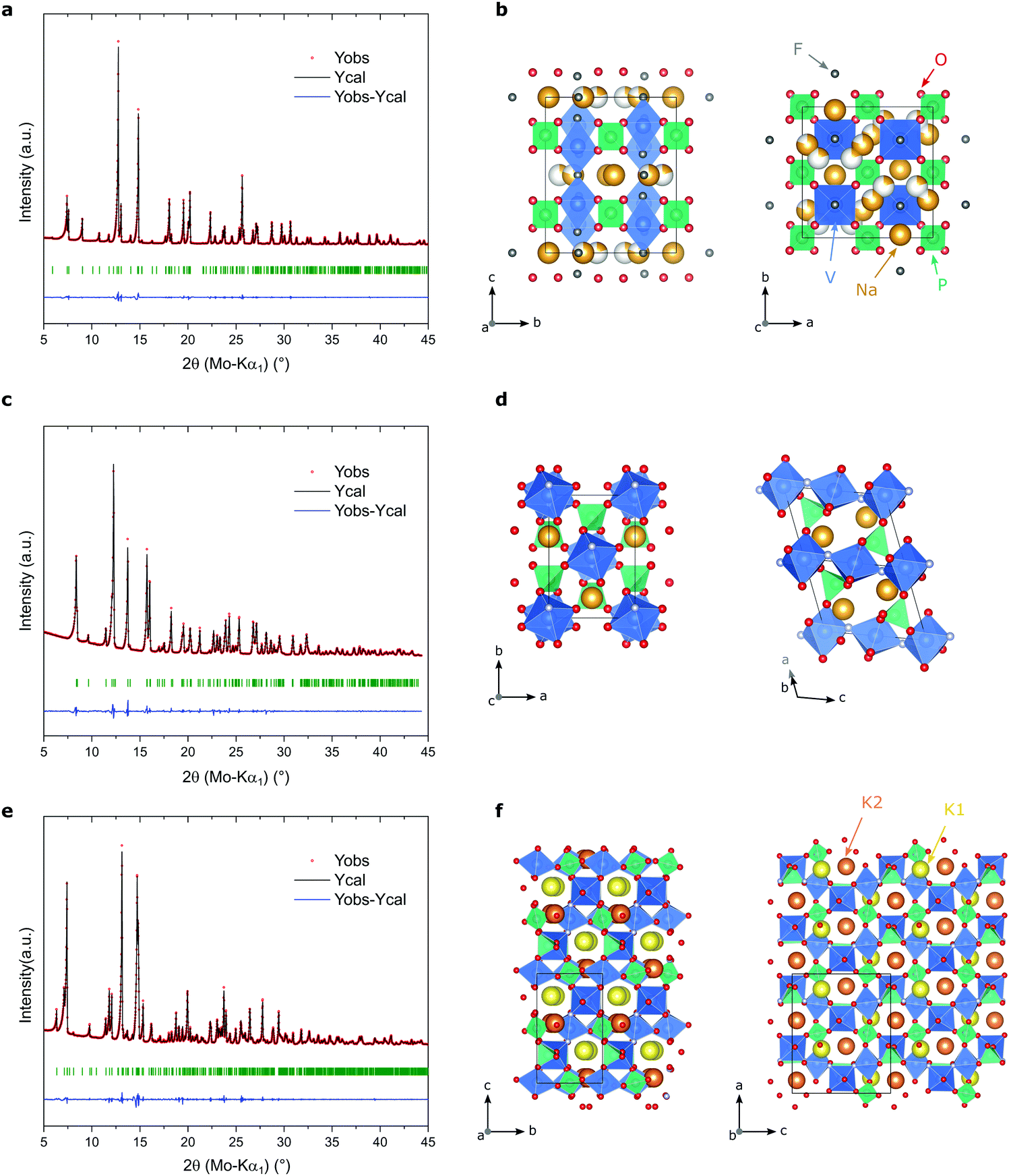 | ||
| Fig. 6 (a, c and e) Experimental and calculated powder XRD patterns of Na3V2(PO4)2F3, NaVPO4F and KVPO4F, respectively, using Le Bail refinement. Experimental data (red dots), calculated diffractograms (black lines), difference curves (blue) and Bragg positions (green) are shown. (b, d and f) Schematic representations of the respective crystallographic structures with VO4F2 octahedra in blue, PO4 tetrahedra in turquoise and alkaline metals in yellow/orange. | ||
As such, the lattice parameters of Na3V2(PO4)2F3 were refined in the orthorhombic Amam space group, as it was recently demonstrated by Bianchini et al.51 to be the more accurate than the widely accepted tetragonal P42/mnm space group, previously proposed by Le Meins et al.8 In this three-dimensional structure (Fig. 6b), vanadium occupies the center of VO4F2 octahedra that are bridged together by one fluorine atom, forming bioctahedra V2O8F3 units. These V2O8F3 units are linked by PO4 tetrahedra, thus creating large tunnels in the [110] and [10] directions where the sodium ions are placed and can quickly migrate, making this material highly relevant for Na-ion battery applications.
The diffractogram of NaVPO4F was refined in the monoclinic framework (space group C2/c) recently described by Boivin et al.11 As can be seen in Fig. 6d, the structure of NaVPO4F closely resembles to that of LiVPO4F (see Fig. 1b for comparison). This tavorite-like framework consists in chains of VO4F2 octahedra in the [001] direction that are bridged together by fluorine atoms. The connection between these chains are made by PO4 tetrahedra, forming channels along the [110] and [10] directions. However, using a simulation method based on the bond valence theory, Boivin and colleagues11 show that the diffusion of sodium ions in this structure is very limited, which explains the low electrochemical performance that they obtained.
KVPO4F was indexed in the well-known orthorhombic KTP framework, as previously reported.12,31,52 In this three-dimensional structure, zigzag chains of fluorine-sharing VO4F2 octahedra are linked together by PO4 tetrahedra. Contrary to the other materials reported herein, the fluorine atoms occupy alternatively equatorial and axial positions in the VO4F2 octahedra. Potassium ions occupy the tunnels along the [100] and [010] directions, the two different sites being denoted in Fig. 6f as K1 and K2, respectively.
The refined lattice parameters of all the synthesized samples are presented in Table 1 and are in good agreement with previous reports.11,12,51,52
| Compound | Space group | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | V (Å3) |
|---|---|---|---|---|---|---|---|---|
| LiVPO4F |
P |
5.1699(1) | 5.3176(1) | 7.4729(1) | 112.645(1) | 112.390(1) | 82.124(1) | 175.30(4) |
| Na3V2(PO4)2F3 | Amam | 9.0432(2) | 9.0505(2) | 10.7686(2) | 90 | 90 | 90 | 881.36(4) |
| NaVPO4F | C2/c | 6.5791(2) | 8.4417(2) | 7.3920(2) | 90 | 117.565(1) | 90 | 363.94(4) |
| KVPO4F | Pna21 | 12.8576(3) | 6.3982(2) | 10.6442(2) | 90 | 90 | 90 | 875.66(5) |
Electrochemistry of Na3V2(PO4)2F3, NaVPO4F, and KVPO4F material
The electrochemical behavior of the Na and K-containing materials was also characterized and the galvanostatic curves obtained are depicted in Fig. 7 and Fig. S4 (ESI†). As stated in the experimental section, NaVPO4F and Na3V2(PO4)2F3 materials were tested in Na-ion half-cells and KVPO4F in a Li-ion half-cell, as reported by Fedotov et al.12 Overall, the galvanostatic profiles of the materials are consistent with previous reports.11,12,14,53 | ||
| Fig. 7 Galvanostatic profiles of (a) Na3V2(PO4)2F3 at C/10 rate and (c) KVPO4F at C/5 rate. (b, d) Discharge capacity retention of both materials at different charge/discharge rates. | ||
As reported by Boivin et al.,11 almost no electrochemical activity could be detected in the tavorite-like NaVPO4F material, even at low C/10 rate (see Fig. S4, ESI†), which is attributed to the very limited Na-ion diffusion in the structure of the material.
On the contrary, at low rate (C/10, see Fig. 7a), a capacity of 120 mA h g−1 is obtained for the Na3V2(PO4)2F3 material, corresponding to the deintercalation and reintercalation of almost 2 sodium ions (1.88 Na+ per formula unit), occurring around 3.65 and 4.18 V vs. Na+/Na. The rate capability of Na3V2(PO4)2F3 was evaluated (Fig. 7b) and proves to be lower than previous reports involving carbon-coated or carbon-based composite materials,14,54 with almost 40% of the initial capacity (at C/10 rate) lost at 1C rate.
The same general observations can be made for the Li-intercalation/deintercalation in KxVPO4F. The first charge of the initial KVPO4F material was performed at C/10 rate up to 4.8 V vs. Li+/Li and this potential was kept for 5 h to ensure a full oxidation of the material. As depicted in (Fig. S5, ESI†), two main features can be seen in the galvanostatic curves. A first plateau, centered around 4.05 V (from ca. 3.8 to 4.3 V vs. Li+/Li), originates from the deintercalation of K+ ions.12 This reaction leads to a specific capacity of 125 mA h g−1, which translates into ca. 0.95 K+ per formula unit. Then, a second plateau occurs from 4.65 V vs. Li+/Li and is attributed to the oxidation of the electrolyte. During the discharge, the material displays a very similar response as obtained by Fedotov et al.,12 with the intercalation of Li+ occurring between 4.3 and 3.7 V vs. Li+/Li (Fig. S4 (ESI†) and Fig. 7c). As for the other obtained phosphate fluoride materials, the high-rate behavior of (K, Li)VPO4F (Fig. 7d) is not as good as previous reports, with only 50% of the initial capacity retained at 1C rate. The high capacity loss observed between C/10 and C/5 rate is partially due to the absence of potentiostatic step at 4.8 V vs. Li+/Li (performed only during the first cycles at C/10 rate). The poor high-rate performance of the reported KVPO4F compound should however be mitigated, as previous reports were made on a carbon-coated material12 and involved the transfer of the oxidized KxVPO4F material in a fresh Li+-containing electrolyte (at the end of the first charge), a step that wasn’t performed herein.
Conclusion
Soft chemistry routes are highly relevant to the synthesis of battery-type materials as they can allow one to obtain quickly and at low temperature highly crystalline materials with a better controlled morphology, as compared to conventional solid-state methods. We report herein a new one-pot fluorolytic route to various crystalline and phase-pure vanadium phosphate fluoride battery-type materials. The use of such a low temperature (240 °C) solution-based method allowed the prevention of both the evaporation of volatile fluorine compounds and the partial oxidation of V(III) in V(IV) in the samples, issues that are commonly faced by solid-state syntheses. As such, a vanadyl defect-free LiVPO4F material could be obtained at a mild temperature for the first time and exhibited a high specific capacity of 147 mA h g−1 at C/10 rate. The tuning of the synthesis parameters allowed us to similarly obtain the related NaVPO4F, Na3V2(PO4)2F3 and KVPO4F materials. While the high-rate performance of the synthesized compounds is below expectations, it could be greatly enhanced by a further optimization with the addition of either a carbon source in the precursor solution or a carbon-coating at the surface of the particles. Indeed, this attribute is inherent to materials synthesized by the widely used solid-state carbothermal route, and proved to considerably improve their power performance.21,54 Additionally, the demonstrated versatility of the reported fluorolytic synthesis route is worth exploring for other fluorine-containing polyanionic systems that are highly relevant for energy storage applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially funded by the Deutsche Forschungsgemeinschaft DFG KE 489/21-1 or PI 762/5-1. Andrea Martin is acknowledged for experimental support and fruitful discussions.References
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- C. Masquelier and L. Croguennec, Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS.
- A. Manthiram and J. B. Goodenough, Lithium insertion into Fe2(MO4)3 frameworks: Comparison of M = W with M = Mo, J. Solid State Chem., 1987, 71, 349–360 CrossRef CAS.
- A. Manthiram and J. B. Goodenough, Lithium insertion into Fe2(SO4)3 frameworks, J. Power Sources, 1989, 26, 403–408 CrossRef CAS.
- W. H. Baur, Die Kristallstruktur des Edelamblygonits LiAlPO4(OH, F), Acta Crystallogr., 1959, 12, 988–994 CrossRef.
- E. L. Belokoneva, O. V. Yakubovich, V. G. Tsirel'son and V. S. Urusov, The refined crystal structure and electronic structure of the nonlinear crystal potassium iron fluorophosphate (KFeFPO4) – a structural analog of potassium titanium oxide phosphate (KTiOPO4), Inorg. Mater., 1990, 26, 595–601 CAS.
- O. V. Yakubovich, O. V. Karimova and O. K. Mel'nikov, The Mixed Anionic Framework in the Structure of Na2{MnF[PO4]}, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 395–397 CrossRef.
- J. M. Le Meins, M. P. Crosnier-Lopez, A. Hemon-Ribaud and G. Courbion, Phase Transitions in the Na3M2(PO4)2F3 Family (M = Al3+, V3+, Cr3+, Fe3+, Ga3+): Synthesis, Thermal, Structural, and Magnetic Studies, J. Solid State Chem., 1999, 148, 260–277 CrossRef CAS.
- S. C. Yin, P. S. Herle, A. Higgins, N. J. Taylor, Y. Makimura and L. F. Nazar, Dimensional Reduction: Synthesis and Structure of Layered Li5M(PO4)2F2 (M = V, Cr), Chem. Mater., 2006, 18, 1745–1752 CrossRef CAS.
- J. Barker, M. Y. Saidi and J. L. Swoyer, Electrochemical Insertion Properties of the Novel Lithium Vanadium Fluorophosphate, LiVPO4F, J. Electrochem. Soc., 2003, 150, A1394–A1398 CrossRef CAS.
- E. Boivin, J.-N. Chotard, T. Bamine, D. Carlier, P. Serras, V. Palomares, T. Rojo, A. Iadecola, L. Dupont, L. Bourgeois, F. Fauth, C. Masquelier and L. Croguennec, Vanadyl-type defects in Tavorite-like NaVPO4F: from the average long range structure to local environments, J. Mater. Chem. A, 2017, 5, 25044–25055 RSC.
- S. S. Fedotov, N. R. Khasanova, A. S. Samarin, O. A. Drozhzhin, D. Batuk, O. M. Karakulina, J. Hadermann, A. M. Abakumov and E. V. Antipov, AVPO4F (A = Li, K): A 4 V Cathode Material for High-Power Rechargeable Batteries, Chem. Mater., 2016, 28, 411–415 CrossRef CAS.
- R. K. B. Gover, A. Bryan, P. Burns and J. Barker, The electrochemical insertion properties of sodium vanadium fluorophosphate, Na3V2(PO4)2F3, Solid State Ionics, 2006, 177, 1495–1500 CrossRef CAS.
- A. Ponrouch, R. Dedryvère, D. Monti, A. E. Demet, J. M. Ateba Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacín, Towards high energy density sodium ion batteries through electrolyte optimization, Energy Environ. Sci., 2013, 6, 2361–2369 RSC.
- H. Huang, T. Faulkner, J. Barker and M. Y. Saidi, Lithium metal phosphates, power and automotive applications, J. Power Sources, 2009, 189, 748–751 CrossRef CAS.
- F. Zhou, X. Zhao and J. R. Dahn, Reactivity of charged LiVPO4F with 1 M LiPF6 EC:DEC electrolyte at high temperature as studied by accelerating rate calorimetry, Electrochem. Commun., 2009, 11, 589–591 CrossRef CAS.
- J.-M. Ateba Mba, C. Masquelier, E. Suard and L. Croguennec, Synthesis and Crystallographic Study of Homeotypic LiVPO4F and LiVPO4O, Chem. Mater., 2012, 24, 1223–1234 CrossRef CAS.
- J. Wang, X. Li, Z. Wang, H. Guo, Y. Zhang, X. Xiong and Z. He, Synthesis and characterization of LiVPO4F/C using precursor obtained through a soft chemical route with mechanical activation assist, Electrochim. Acta, 2013, 91, 75–81 CrossRef CAS.
- J.-c. Zheng, B. Zhang and Z.-H. Yang, Novel synthesis of LiVPO4F cathode material by chemical lithiation and postannealing, J. Power Sources, 2012, 202, 380–383 CrossRef CAS.
- P. F. Xiao, M. O. Lai and L. Lu, Transport and electrochemical properties of high potential tavorite LiVPO4F, Solid State Ionics, 2013, 242, 10–19 CrossRef CAS.
- M. Kim, S. Lee and B. Kang, Fast-Rate Capable Electrode Material with Higher Energy Density than LiFePO4: 4.2 V LiVPO4F Synthesized by Scalable Single-Step Solid-State Reaction, Adv. Sci., 2016, 3, 1500366 CrossRef.
- J. Barker, R. K. B. Gover, P. Burns, A. Bryan, M. Y. Saidi and J. L. Swoyer, Performance Evaluation of Lithium Vanadium Fluorophosphate in Lithium Metal and Lithium-Ion Cells, J. Electrochem. Soc., 2005, 152, A1776–A1779 CrossRef CAS.
- E. Kemnitz, U. Groß, S. Rüdiger and C. S. Shekar, Amorphous Metal Fluorides with Extraordinary High Surface Areas, Angew. Chem., Int. Ed., 2003, 42, 4251–4254 CrossRef CAS.
- J. Noack, K. Scheurell, E. Kemnitz, P. Garcia-Juan, H. Rau, M. Lacroix, J. Eicher, B. Lintner, T. Sontheimer, T. Hofmann, J. Hegmann, R. Jahn and P. Löbmann, MgF2 antireflective coatings by sol–gel processing: film preparation and thermal densification, J. Mater. Chem., 2012, 22, 18535–18541 RSC.
- A. Rehmer, K. Scheurell and E. Kemnitz, Formation of nanoscopic CaF2via a fluorolytic sol–gel process for antireflective coatings, J. Mater. Chem. C, 2015, 3, 1716–1723 RSC.
- Y. Guo, P. Gaczyński, K.-D. Becker and E. Kemnitz, Sol–Gel Synthesis and Characterisation of Nanoscopic FeF3-MgF2 Heterogeneous Catalysts with Bi-Acidic Properties, ChemCatChem, 2013, 5, 2223–2232 CrossRef CAS.
- A. Rehmer, K. Scheurell, G. Scholz and E. Kemnitz, Sol–Gel-Synthesis of Nanoscopic Complex Metal Fluorides, Nanomaterials, 2017, 7, 362 CrossRef.
- A. Martin, M.-L. Doublet, E. Kemnitz and N. Pinna, Reversible Sodium and Lithium Insertion in Iron Fluoride Perovskites, Adv. Funct. Mater., 2018, 28, 1802057 CrossRef.
- T. Krahl, F. M. Winkelmann, A. Martin, N. Pinna and E. Kemnitz, Novel Synthesis of Anhydrous and Hydroxylated CuF2 Nanoparticles and Their Potential for Lithium Ion Batteries, Chem. – Eur. J., 2018, 24, 7177–7187 CrossRef CAS.
- N. Goubard-Bretesché, E. Kemnitz and N. Pinna, Fluorolytic Sol–Gel Route and Electrochemical Properties of Polyanionic Transition-Metal Phosphate Fluorides, Chem. – Eur. J., 2019, 25, 6189–6195 CrossRef.
- K. Chihara, A. Katogi, K. Kubota and S. Komaba, KVPO4F and KVOPO4 toward 4 volt-class potassium-ion batteries, Chem. Commun., 2017, 53, 5208–5211 RSC.
- S. Rudiger and E. Kemnitz, The fluorolytic sol–gel route to metal fluorides-a versatile process opening a variety of application fields, Dalton Trans., 2008, 1117–1127 RSC.
- E. Kemnitz and J. Noack, The non-aqueous fluorolytic sol–gel synthesis of nanoscaled metal fluorides, Dalton Trans., 2015, 44, 19411–19431 RSC.
- J. Livage, P. Barboux, M. T. Vandenborre, C. Schmutz and F. Taulelle, Sol–gel synthesis of phosphates, J. Non-Cryst. Solids, 1992, 147–148, 18–23 CrossRef CAS.
- M.-G. Willinger, G. Clavel, W. Di and N. Pinna, A general soft-chemistry route to metal phosphate nanocrystals, J. Ind. Eng. Chem., 2009, 15, 883–887 CrossRef CAS.
- T. Mueller, G. Hautier, A. Jain and G. Ceder, Evaluation of Tavorite-Structured Cathode Materials for Lithium-Ion Batteries Using High-Throughput Computing, Chem. Mater., 2011, 23, 3854–3862 CrossRef CAS.
- B. L. Ellis, T. N. Ramesh, L. J. M. Davis, G. R. Goward and L. F. Nazar, Structure and Electrochemistry of Two-Electron Redox Couples in Lithium Metal Fluorophosphates Based on the Tavorite Structure, Chem. Mater., 2011, 23, 5138–5148 CrossRef CAS.
- M. L. Lindberg and W. T. Pecora, Tavorite and barbosalite, two new phosphate minerals from Minas Gerais, Brazil, Am. Mineral., 1955, 40, 952–966 CAS.
- N. Marx, L. Croguennec, D. Carlier, A. Wattiaux, F. L. Cras, E. Suard and C. Delmas, The structure of tavorite LiFePO4(OH) from diffraction and GGA + U studies and its preliminary electrochemical characterization, Dalton Trans., 2010, 39, 5108–5116 RSC.
- T. N. Ramesh, K. T. Lee, B. L. Ellis and L. F. Nazar, Tavorite Lithium Iron Fluorophosphate Cathode Materials: Phase Transition and Electrochemistry of LiFePO4F–Li2FePO4F, Electrochem. Solid-State Lett., 2010, 13, A43–A47 CrossRef CAS.
- E. Boivin, J.-N. Chotard, M. Ménétrier, L. Bourgeois, T. Bamine, D. Carlier, F. Fauth, E. Suard, C. Masquelier and L. Croguennec, Structural and electrochemical studies of a new Tavorite composition: LiVPO4OH, J. Mater. Chem. A, 2016, 4, 11030–11045 RSC.
- T. A. Kerr, J. Gaubicher and L. F. Nazar, Highly Reversible Li Insertion at 4 V in ε-VOPO4/α-LiVOPO4 Cathodes, Electrochem. Solid-State Lett., 2000, 3, 460–462 CrossRef CAS.
- E. Boivin, J.-N. Chotard, M. Ménétrier, L. Bourgeois, T. Bamine, D. Carlier, F. Fauth, C. Masquelier and L. Croguennec, Oxidation under Air of Tavorite LiVPO4F: Influence of Vanadyl-Type Defects on Its Electrochemical Properties, J. Phys. Chem. C, 2016, 120, 26187–26198 CrossRef CAS.
- M. Kim, S. Lee and B. Kang, High Energy Density Polyanion Electrode Material: LiVPO4O1−xFx (x ≈ 0.25) with Tavorite Structure, Chem. Mater., 2017, 29, 4690–4699 CrossRef CAS.
- K. L. Harrison and A. Manthiram, Microwave-Assisted Solvothermal Synthesis and Characterization of Various Polymorphs of LiVOPO4, Chem. Mater., 2013, 25, 1751–1760 CrossRef CAS.
- M. Niederberger and N. Pinna, Metal oxide nanoparticles in organic solvents: synthesis, formation, assembly and application, Springer, Dordrecht, 2009 Search PubMed.
- N. Pinna, M. Antonietti and M. Niederberger, A novel nonaqueous route to V2O3 and Nb2O5 nanocrystals, Colloids Surf., A, 2004, 250, 211–213 CrossRef CAS.
- E. Boivin, R. David, J.-N. Chotard, T. Bamine, A. Iadecola, L. Bourgeois, E. Suard, F. Fauth, D. Carlier, C. Masquelier and L. Croguennec, LiVPO4F1–yOy Tavorite-Type Compositions: Influence of the Concentration of Vanadyl-Type Defects on the Structure and Electrochemical Performance, Chem. Mater., 2018, 30, 5682–5693 CrossRef CAS.
- M. Bianchini, J. M. Ateba-Mba, P. Dagault, E. Bogdan, D. Carlier, E. Suard, C. Masquelier and L. Croguennec, Multiple phases in the ε-VPO4O–LiVPO4O–Li2VPO4O system: a combined solid state electrochemistry and diffraction structural study, J. Mater. Chem. A, 2014, 2, 10182–10192 RSC.
- M. Wagemaker and F. M. Mulder, Properties and Promises of Nanosized Insertion Materials for Li-Ion Batteries, Acc. Chem. Res., 2013, 46, 1206–1215 CrossRef CAS.
- M. Bianchini, N. Brisset, F. Fauth, F. Weill, E. Elkaim, E. Suard, C. Masquelier and L. Croguennec, Na3V2(PO4)2F3 Revisited: A High-Resolution Diffraction Study, Chem. Mater., 2014, 26, 4238–4247 CrossRef CAS.
- H. Kim, D.-H. Seo, M. Bianchini, R. J. Clément, H. Kim, J. C. Kim, Y. Tian, T. Shi, W.-S. Yoon and G. Ceder, A New Strategy for High-Voltage Cathodes for K-Ion Batteries: Stoichiometric KVPO4F, Adv. Energy Mater., 2018, 8, 1801591 CrossRef.
- K. Chihara, A. Kitajou, I. D. Gocheva, S. Okada and J.-I. Yamaki, Cathode properties of Na3M2(PO4)2F3 [M = Ti, Fe, V] for sodium-ion batteries, J. Power Sources, 2013, 227, 80–85 CrossRef CAS.
- C. Zhu, C. Wu, C.-C. Chen, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, A High Power–High Energy Na3V2(PO4)2F3 Sodium Cathode: Investigation of Transport Parameters, Rational Design and Realization, Chem. Mater., 2017, 29, 5207–5215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00325h |
| This journal is © the Partner Organisations 2019 |