
Drawing a clear mechanistic picture for the aggregation-induced emission process†
Haoke
Zhang‡
 abc,
Junkai
Liu‡
abc,
Lili
Du
de,
Chao
Ma
f,
Nelson L. C.
Leung
abc,
Yingli
Niu
g,
Anjun
Qin
h,
Jingzhi
Sun
i,
Qian
Peng
j,
Herman H. Y.
Sung
a,
Ian D.
Williams
a,
Ryan T. K.
Kwok
abc,
Jacky W. Y.
Lam
abc,
Kam Sing
Wong
f,
David Lee
Phillips
d and
Ben Zhong
Tang
*abch
abc,
Junkai
Liu‡
abc,
Lili
Du
de,
Chao
Ma
f,
Nelson L. C.
Leung
abc,
Yingli
Niu
g,
Anjun
Qin
h,
Jingzhi
Sun
i,
Qian
Peng
j,
Herman H. Y.
Sung
a,
Ian D.
Williams
a,
Ryan T. K.
Kwok
abc,
Jacky W. Y.
Lam
abc,
Kam Sing
Wong
f,
David Lee
Phillips
d and
Ben Zhong
Tang
*abch
aDepartment of Chemistry, The Hong Kong University of Science and Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
bThe Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, HKUST, Clear Water Bay, Kowloon, Hong Kong, China
cHKUST-Shenzhen Research Institute, No. 9 Yuexing 1st RD, South Area, Hi-tech Park Nanshan, Shenzhen 518057, China
dDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
eInstitute of Life Sciences, Jiangsu University, Zhenjiang 212013, China
fDepartment of Physics, HKUST, Clear Water Bay, Kowloon, Hong Kong, China
gDepartment of Physics, School of Science, Beijing Jiaotong University, Shangyuancun No. 3, Beijing 100044, China
hCenter for Aggregation-Induced Emission, SCUT-HKUST Joint Research Institute, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
iMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
jKey Laboratory of Organic Solids, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 17th April 2019
Abstract
Fluorescence is a widely used method to monitor many biological and abiological processes as well as being famous for its use in display devices. Currently, its wavelength can be accurately tuned in organic fluorophores by regulating their conjugation length. However, emission efficiency is still an experiential parameter. In this work, we investigate the photophysical properties of two stilbene-based isomerides. The different positions of one methyl group make di-o-methyl substituted 2,4,6-TMe-DPE more twisted than mono-o-methyl substituted 2,4,5-TMe-DPE. Experimental results show that the twisted structure is nonluminescent in solution and exhibits a typical aggregation-induced emission effect. However, the other planar isomer shows strong emission both in solution and in aggregate states. Steady-state and time-resolved spectroscopy, and quantum mechanical calculation suggest that nonradiative decay is predominant in 2,4,6-TMe-DPE and that excited-state double-bond torsion contributes a lot to the nonradiative decay channel. This work draws a clear photophysical picture for the aggregation-induced emission process and enables the luminescent behavior to become predictable and controllable.
Introduction
Luminescence is a natural phenomenon and has been observed for thousands of years, as recorded in a Chinese book in 1500 B.C.1 Meanwhile, it is important for life and our society, playing a great role in scientific discovery and technological innovation.2 Reviewing its history, in 1852, Sir George Gabriel Stokes published a landmark article entitled “On the Change of Refrangibility of Light” to describe how visible blue light was generated from ultraviolet light.3 He named this phenomenon fluorescence. The next year Stokes discovered another interesting phenomenon in platinocyanide.4 This metal complex showed strong emission in the solid state, but its solution looks like mere water showing no fluorescence. One century later, in 1954, Th. Förster reported the opposite effect. He observed that the strong emission of pyrene in dilute solution was quenched with an increase in solution concentration.5 Further studies found that some compounds were non-emissive in solution but emitted intensively in the aggregate state, while others showed strong fluorescence as single molecular species but no light in aggregates.6–8 These phenomena have certainly been observed by many researchers for a long time. However, they were thrown into a corner as chemists and physicists treated them as common features, unworthy of intensive investigation.In 2001, Tang et al. occasionally found that the highly emissive solid powders of 1-methyl-1,2,3,4,5-pentaphenylsilole became nonluminescent when dissolved in good solvents.9 However, they did not ignore this phenomenon this time as had been done before. Tang et al. conceptually coined the phenomenon of aggregation-induced emission (AIE), although the underlying mechanism remained unclear. In the past seventeen years, many scientists all over the world have stepped into this arena to investigate the mechanisms and applications of AIE.10 Currently, most developed AIE luminogens (AIEgens) show twisted structures and restriction in intramolecular motion (RIM) has been proposed as its working mechanism.11,12 In contrast, some planar molecules often show an aggregation-caused quenching (ACQ) effect attributed to excimer formation.6,13 Actually, knowledge of the photophysical processes of AIE and ACQ is still limited and superficial because of the molecular diversity and complicated aggregation processes.14–22
It is never easy to draw a big and clear picture of fluorescence for all kinds of chromophores. In this work, going back to the old question, we try to clarify why some molecules are emissive in solution but others are nonluminescent. Two kinds of fluorophores are selected: twisted structures with an AIE effect and planar molecules. As shown in Fig. 1A and B, (E)-1,2-bis(2,4,5-trimethylphenyl)ethene (2,4,5-TMe-DPE) adopted a planar structure and its fluorescence quantum yield in tetrahydrofuran (THF) solution (ΦF,solution) reached 13.4%. However, its isomeride (E)-1,2-bis(2,4,6-trimethylphenyl)ethene (2,4,6-TMe-DPE) with a twisted structure and a larger dihedral angle (α) of 41.7° showed almost no emission. Its ΦF,solution was as low as 0.6%. Steady-state and time-resolved spectroscopy measurements, and theoretical calculation data suggested that excited-state “double-bond” torsion (ESDBT) contributed greatly to nonradiative decay in solution. When the double bond was replaced by other groups, such as a benzene ring, the resulting molecules still had a twisted structure but were more emissive in solution due to the partial suppression of nonradiative decay. These results demonstrated that the ESDBT process played an important role in the nonluminescent feature of vinyl-based AIEgens, and could serve as a guide to control fluorescence behavior.
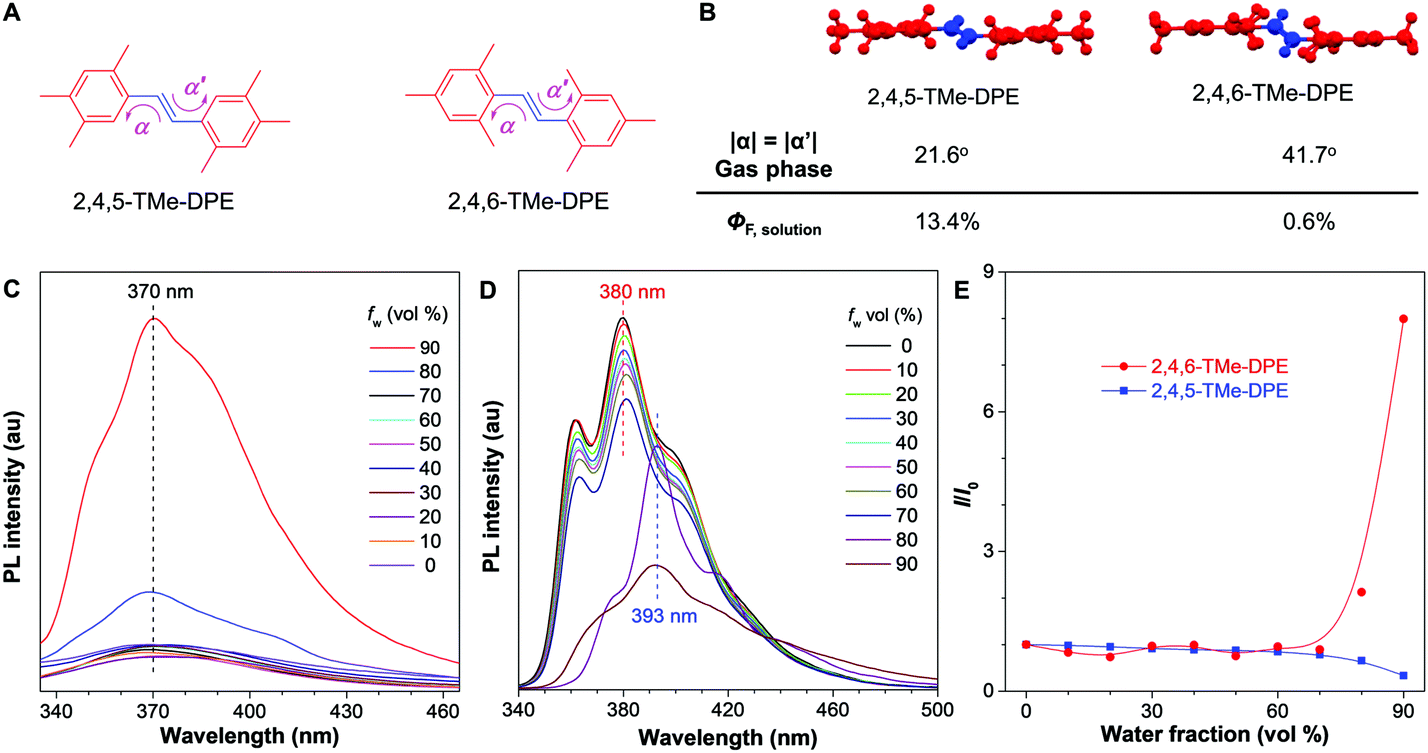 | ||
| Fig. 1 Photophysical properties of 2,4,6-TMe-DPE and 2,4,5-TMe-DPE. (A) Structures of 2,4,5-TMe-DPE and 2,4,6-TMe-DPE. (B) Conformation in the gas phase calculated by DFT B3LYP/6-311G** and absolute fluorescence quantum yield of solution (ΦF) measured in a THF solution. PL spectra of (C) 2,4,6-TMe-DPE and (D) 2,4,5-TMe-DPE in THF/water mixtures with different water fractions (fw). (E) Plots of relative PL intensity (I/I0) of 2,4,6-TMe-DPE and 2,4,5-TMe-DPE versus the composition of their THF/water mixture. Concentration = 10 μM, λex = 280 nm, I0 = PL intensity at fw = 0%. | ||
Results and discussion
(A) Synthesis, characterization and photophysical properties
2,4,5-TMe-DPE, 2,4,6-TMe-DPE and other relevant molecules were designed and synthesized in this project according to Fig. S1 in the ESI.† All the molecules were characterized and confirmed by NMR and mass spectroscopy (Fig. S2–S19, ESI†). Suitable crystals were grown from their solutions and characterized crystallographically (Fig. S20–S23, ESI†). As shown in Fig. 1B, two isomerides of 2,4,5-TMe-DPE and 2,4,6-TMe-DPE showed quite different conformations. Ground-state optimization performed in the gas phase indicated that the di-o-methyl substituted 2,4,6-TMe-DPE had a twisted structure with α = 41.7°, but a more planar structure was observed in mono-o-methyl substituted 2,4,5-TMe-DPE with α = 21.6°. The crystal structure showed α of 54° and 27° for 2,4,6-TMe-DPE and 2,4,5-TMe-DPE, respectively, which were close to the values obtained from gas-phase optimization. UV-vis absorption spectra indicated that the maximum absorption wavelength (λabs) of 2,4,5-TMe-DPE was 30 nm longer than that of 2,4,6-TMe-DPE (Fig. S24, ESI†), which suggested that the planar 2,4,5-TMe-DPE had a better electronic conjugation than the twisted 2,4,6-TMe-DPE.2,23Inspired by their different values of ΦF,solution, we recorded the steady-state photoluminescence (PL) spectra of these two compounds in THF/water mixtures with different water fractions (fw). Fig. 1C shows that almost no emission was observed in 2,4,6-TMe-DPE when fw was lower than 80%. However, a dramatic PL enhancement was detected at fw ≥ 80% due to the formation of aggregates,24 which showed an archetypical AIE characteristic (Fig. 1E). In contrast, 2,4,5-TMe-DPE already exhibited strong emission in pure THF solution (Fig. 1D). From fw = 0 to 70%, Fig. 1E indicates that the PL intensity (I) of 2,4,5-TMe-DPE at 380 nm showed almost no change. A slight decrease in I and a 13 nm bathochromic shift in the maximum emission wavelength (λem) were observed when the aggregates formed at fw ≥ 80%. According to the recorded data in Fig. 1, 2,4,5-TMe-DPE and 2,4,6-TMe-DPE are suitable candidates to answer our question – “why are twisted molecules nonluminescent in solution?”
In addition to that, the aggregation-induced bathochromic shift of λem in 2,4,5-TMe-DPE also arrested our attention, as no wavelength shift was observed in 2,4,6-TMe-DPE. Not only the excited-state performance, but the absorption spectra at both fw = 0 and 90% were also checked. Fig. S24 (ESI†) shows that a 33 nm red shift of λabs was observed in 2,4,5-TMe-DPE when its water fraction increased from 0 to 90%, but only a 5 nm difference was found in 2,4,6-TMe-DPE. To decipher the cause, both intra- and intermolecular factors were taken into consideration for calculation. The rotational barriers for ground-state intramolecular motion are shown in Fig. 2A. All of the calculations started from their lowest-energy conformation. Then, torsion angles α = −α′ decreased or increased simultaneously. A planar structure was formed when the value of α reached 0°. At α = 90°, the surrounding phenyl rings were perpendicular to the core. Generally, planar structures exhibited narrower electronic band gaps than perpendicular ones due to better electronic delocalization.25Fig. 2A suggests that a low rotational barrier (<5 kJ mol−1) was needed for 2,4,5-TMe-DPE to form planar structures. However, 2,4,6-TMe-DPE needed a much higher energy (around 100 kJ mol−1) to undergo planarization. It is easy to conclude that the aggregation-induced planarization in 2,4,5-TMe-DPE could be one reason for the bathochromic shift of λabs and λem.26 Meanwhile, crystal packing structures suggested that J-aggregates were formed in both 2,4,5-TMe-DPE and 2,4,6-TMe-DPE (Fig. S25 and S26, ESI†).27,28 However, theoretical calculation on their packing structures showed that 2,4,5-TMe-DPE had a stronger dimer interaction than 2,4,6-TMe-DPE (Fig. 2B, C and Fig. S27, ESI†). From monomer to dimer, Fig. 2B suggests that the energy gap (Eg) of 2,4,6-TMe-DPE decreased from 5.354 to 5.291 eV, a small decrease of 0.063 eV. However, a 0.166 eV decrease was observed in planar 2,4,5-TMe-DPE (Fig. 2C), which was almost three times bigger than that of twisted 2,4,6-TMe-DPE. Meanwhile, by comparison with 2,4,6-TMe-DPE, a larger intermolecular overlap of the electron cloud at the lowest unoccupied molecular orbital was observed in 2,4,5-TMe-DPE, which also suggested that planar 2,4,5-TMe-DPE showed a stronger intermolecular through-space conjugation than 2,4,6-TMe-DPE when their dimers were formed in the aggregate state.29–31 Therefore, apart from a planarity effect, formation of a dimer in the aggregate state also contributed a lot to the bathochromic shift of λem and λabs in 2,4,5-TMe-DPE.
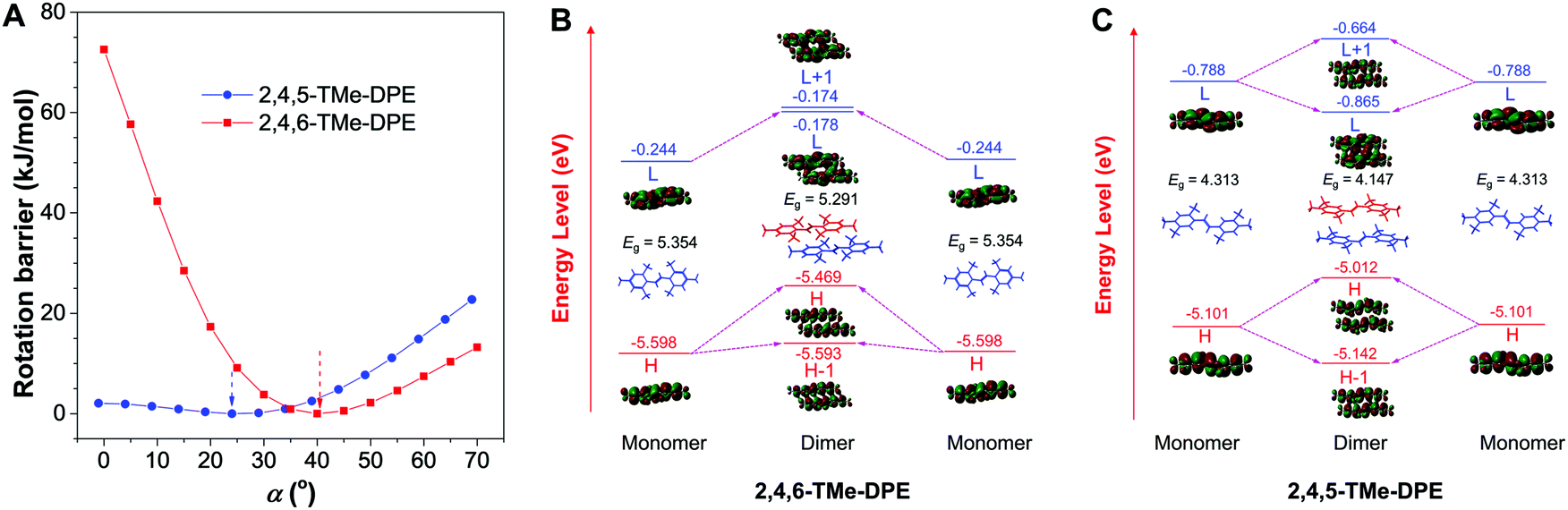 | ||
| Fig. 2 Quantum mechanics simulation on 2,4,5-TMe-DPE and 2,4,6-TMe-DPE. (A) Rotational barrier of 2,4,5-TMe-DPE and 2,4,6-TMe-DPE in the ground state, |α| = |α′|. Calculated energy levels of the monomer and dimer of (B) 2,4,6-TMe-DPE and (C) 2,4,5-TMe-DPE in the ground state calculated by DFT B3LYP/6-31G**. Isovalue = 0.01 e Å−3. H: HOMO (highest occupied molecular orbital), L: LUMO (lowest unoccupied molecular orbital), Eg: energy gap between LUMO and HOMO. The monomer and dimer conformation were extracted from their crystal structures without further optimization. | ||
(B) Femtosecond transient absorption spectroscopy
In order to clarify the photophysical performance of 2,4,5-TMe-DPE and 2,4,6-TMe-DPE, their excited-state properties were investigated further. Generally, the fluorescence quantum yield (ΦF) can be expressed as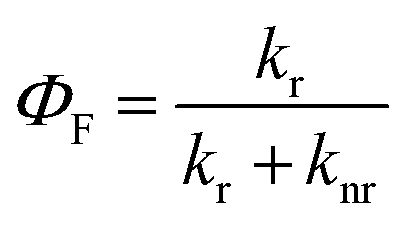 , where kr is the radiative decay rate of fluorescence and knr is the nonradiative decay rate. According to previous studies,32kr and knr could be expressed as ΦF/τ and (1 − ΦF)/τ, respectively, where τ is the excited-state decay lifetime. As a special type of pump–probe technology, femtosecond transient absorption spectroscopy (fs-TA) allows us to monitor the evolution of the molecules in their excited states.33–35 In this work, a 267 nm pump beam with a 120 fs laser pulse was used to excite these two molecules. All the measurements were carried out in THF solution. In Fig. 3, the positive induced optical density (ΔOD) around 600 nm corresponded to the excited-state absorption (ESA) band and the negative ΔOD around 380 nm was due to the stimulated emission (SE) band. Fig. 3A shows the early spectral evolution of 2,4,6-TMe-DPE. The slightly delayed growth of the ESA band reflected an internal conversion of Sn → S1. The evolution from 1.06 to 12.9 ps shown in Fig. 3B was related to an isomerization process.34 Generally, this isomerization process corresponds to excited-state double-bond torsion, double-bond elongation and phenyl ring twisting. The kinetics for 2,4,6-TMe-DPE at 600 nm can be fitted well by a one-exponential equation with a lifetime (τ) of 2.1 ps (Fig. 3C). According to the 0.6% quantum yield in 2,4,6-TMe-DPE, its knr and kr were calculated to be 4.73 × 1011 s−1 and 2.86 × 109 s−1, respectively. Meanwhile, the fs-TA spectra were also recorded for 2,4,5-TMe-DPE (Fig. 3D–F). Unlike the twisted 2,4,6-TMe-DPE, the SE band at 380 nm reached its maximum at 2.12 ps, which indicates that the planar 2,4,5-TMe-DPE showed a longer Sn → S1 relaxation time and a stabilization process before the isomerization occurred. It is noteworthy that two time constants of 45.2 (τ1) and 363 ps (τ2) were found for 2,4,5-TMe-DPE, which indicated that two processes were involved in the excited-state cooling process.34 Previous studies showed that the amplitude-weighted average lifetime τave in a two-time-constant system could be expressed as
, where kr is the radiative decay rate of fluorescence and knr is the nonradiative decay rate. According to previous studies,32kr and knr could be expressed as ΦF/τ and (1 − ΦF)/τ, respectively, where τ is the excited-state decay lifetime. As a special type of pump–probe technology, femtosecond transient absorption spectroscopy (fs-TA) allows us to monitor the evolution of the molecules in their excited states.33–35 In this work, a 267 nm pump beam with a 120 fs laser pulse was used to excite these two molecules. All the measurements were carried out in THF solution. In Fig. 3, the positive induced optical density (ΔOD) around 600 nm corresponded to the excited-state absorption (ESA) band and the negative ΔOD around 380 nm was due to the stimulated emission (SE) band. Fig. 3A shows the early spectral evolution of 2,4,6-TMe-DPE. The slightly delayed growth of the ESA band reflected an internal conversion of Sn → S1. The evolution from 1.06 to 12.9 ps shown in Fig. 3B was related to an isomerization process.34 Generally, this isomerization process corresponds to excited-state double-bond torsion, double-bond elongation and phenyl ring twisting. The kinetics for 2,4,6-TMe-DPE at 600 nm can be fitted well by a one-exponential equation with a lifetime (τ) of 2.1 ps (Fig. 3C). According to the 0.6% quantum yield in 2,4,6-TMe-DPE, its knr and kr were calculated to be 4.73 × 1011 s−1 and 2.86 × 109 s−1, respectively. Meanwhile, the fs-TA spectra were also recorded for 2,4,5-TMe-DPE (Fig. 3D–F). Unlike the twisted 2,4,6-TMe-DPE, the SE band at 380 nm reached its maximum at 2.12 ps, which indicates that the planar 2,4,5-TMe-DPE showed a longer Sn → S1 relaxation time and a stabilization process before the isomerization occurred. It is noteworthy that two time constants of 45.2 (τ1) and 363 ps (τ2) were found for 2,4,5-TMe-DPE, which indicated that two processes were involved in the excited-state cooling process.34 Previous studies showed that the amplitude-weighted average lifetime τave in a two-time-constant system could be expressed as  , where A1 and A2 are the fitting coefficients which were 0.0075 and 0.01626 in 2,4,5-TMe-DPE, respectively.36,37 Based on this formula, the calculated τave was equal to 263 ps. Similarly, from the ΦF,solution of 13.4% in 2,4,5-TMe-DPE, its knr and kr were calculated to be 3.29 × 109 s−1 and 5.10 × 108 s−1, respectively. Time-resolved fluorescence spectroscopy measurement was also carried out for 2,4,5-TMe-DPE and the obtained knr and kr were quite close to the results calculated from fs-TA (Fig. S28, ESI†).
, where A1 and A2 are the fitting coefficients which were 0.0075 and 0.01626 in 2,4,5-TMe-DPE, respectively.36,37 Based on this formula, the calculated τave was equal to 263 ps. Similarly, from the ΦF,solution of 13.4% in 2,4,5-TMe-DPE, its knr and kr were calculated to be 3.29 × 109 s−1 and 5.10 × 108 s−1, respectively. Time-resolved fluorescence spectroscopy measurement was also carried out for 2,4,5-TMe-DPE and the obtained knr and kr were quite close to the results calculated from fs-TA (Fig. S28, ESI†).
 | ||
| Fig. 3 Femtosecond transient absorption (fs-TA) spectra. (A), (B) 2,4,6-TMe-DPE and (D), (E) 2,4,5-TMe-DPE in THF solution with 267 nm excitation. Kinetic traces of (C) 2,4,6-TMe-DPE at 600 nm and (F) 2,4,5-TMe-DPE at 560 nm. The solid lines indicate a fitting of the data using a single exponential function. | ||
According to the above fs-TA results, the twisted 2,4,6-TMe-DPE (knr = 4.73 × 1011 s−1) possessed a faster and easier excited-state molecular motion than the planar 2,4,5-TMe-DPE (knr = 3.29 × 109 s−1), which suggested that nonradiative decay was predominant in the photophysical process of 2,4,6-TMe-DPE. In contrast, the restricted molecular motion in 2,4,5-TMe-DPE made its radiative decay comparable with the nonradiative transitions and that was why a ΦF,solution of 13.4% was observed in this planar isomeride. However, the obtained data can only tell us that the nonluminescent property is caused by the excited-state molecular motion, but what kinds of motions are involved and which are predominant is still unclear.
(C) Quantum mechanical calculation
Theoretical calculation is expected to provide further insight into the excited-state molecular motion. Quantum mechanical (QM) theory of time-dependent density functional theory (TDDFT) was selected to perform the calculation in the excited state. Reorganization energy (λ) reflects the intrinsic geometry change upon photoexcitation, which can be obtained by four-point calculation according to the adiabatic potential energy surface.38 Each λ versus the normal mode wavenumber provides essential details of the contribution to the excited-state deactivation from multiple intramolecular motions.39 Previous studies revealed that normal modes in the low-frequency region were assigned to the twisting motion, and bond stretching corresponded to high-frequency modes. Peng et al. have further pointed out that the low-frequency vibration modes tended to mix with each other upon excitation to activate multiple nonradiative decay pathways. Such multi-mode mixing is known as the Duschinsky rotation effect (DRE), which can greatly increase the nonradiative decay rates.40,41The results of λ for 2,4,6-TMe-DPE and 2,4,5-TMe-DPE are summarized in Fig. 4A and C, respectively. 2,4,6-TMe-DPE showed an enormous λ which reached 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 384 cm−1 and most of them were located in the low-frequency region. As expected, 80.8% of the λ was contributed by the change in dihedral angle. Meanwhile, a 70% change in dihedral angle was caused by ESDBT. However, in 2,4,5-TMe-DPE, the λ was as low as 2883 cm−1 and 69.2% of it was assigned to the change in bond length. The statistical result suggested that a 67% change of bond length was attributable to double-bond elongation. At the same time, the calculated λ for 2,4,6-TMe-DPE and 2,4,5-TMe-DPE in the crystal were 4187 and 2689 cm−1, respectively (Fig. S29, ESI†). Obviously, the ESDBT of 2,4,6-TMe-DPE has been supressed in the aggregate state. In contrast, 2,4,5-TMe-DPE has a similar λ in both gas phase and crystal, which agrees with the experimental results. The demonstration of DRE drew a deeper understanding of the decay pathways. Contour maps of the Duschinsky rotation matrix (DRM) for the lowest 50 normal modes are plotted in Fig. 4B and D. Previous studies pointed out that the more diagonal elements of the DRM deviated from 1 and the off-diagonal elements were non-zero, with more significant DRE and larger nonradiative decay rates.40 A comparison of DRM between 2,4,6-TMe-DPE (Fig. 4B) and 2,4,5-TMe-DPE (Fig. 4D) indicated that 2,4,6-TMe-DPE had more off-diagonal elements. Meanwhile, most of the diagonal elements in 2,4,6-TMe-DPE deviated from 1, which suggested that multiple low-frequency twisting motions contributed a lot to the nonradiative deactivation of excitons. The above results on λ and DRM indicated that in twisted 2,4,6-TMe-DPE, most of the excited-state energy was released nonradiatively by means of ESDBT. However, ESDBT was suppressed in 2,4,5-TMe-DPE and the radiative decay rate increased dramatically.
384 cm−1 and most of them were located in the low-frequency region. As expected, 80.8% of the λ was contributed by the change in dihedral angle. Meanwhile, a 70% change in dihedral angle was caused by ESDBT. However, in 2,4,5-TMe-DPE, the λ was as low as 2883 cm−1 and 69.2% of it was assigned to the change in bond length. The statistical result suggested that a 67% change of bond length was attributable to double-bond elongation. At the same time, the calculated λ for 2,4,6-TMe-DPE and 2,4,5-TMe-DPE in the crystal were 4187 and 2689 cm−1, respectively (Fig. S29, ESI†). Obviously, the ESDBT of 2,4,6-TMe-DPE has been supressed in the aggregate state. In contrast, 2,4,5-TMe-DPE has a similar λ in both gas phase and crystal, which agrees with the experimental results. The demonstration of DRE drew a deeper understanding of the decay pathways. Contour maps of the Duschinsky rotation matrix (DRM) for the lowest 50 normal modes are plotted in Fig. 4B and D. Previous studies pointed out that the more diagonal elements of the DRM deviated from 1 and the off-diagonal elements were non-zero, with more significant DRE and larger nonradiative decay rates.40 A comparison of DRM between 2,4,6-TMe-DPE (Fig. 4B) and 2,4,5-TMe-DPE (Fig. 4D) indicated that 2,4,6-TMe-DPE had more off-diagonal elements. Meanwhile, most of the diagonal elements in 2,4,6-TMe-DPE deviated from 1, which suggested that multiple low-frequency twisting motions contributed a lot to the nonradiative deactivation of excitons. The above results on λ and DRM indicated that in twisted 2,4,6-TMe-DPE, most of the excited-state energy was released nonradiatively by means of ESDBT. However, ESDBT was suppressed in 2,4,5-TMe-DPE and the radiative decay rate increased dramatically.
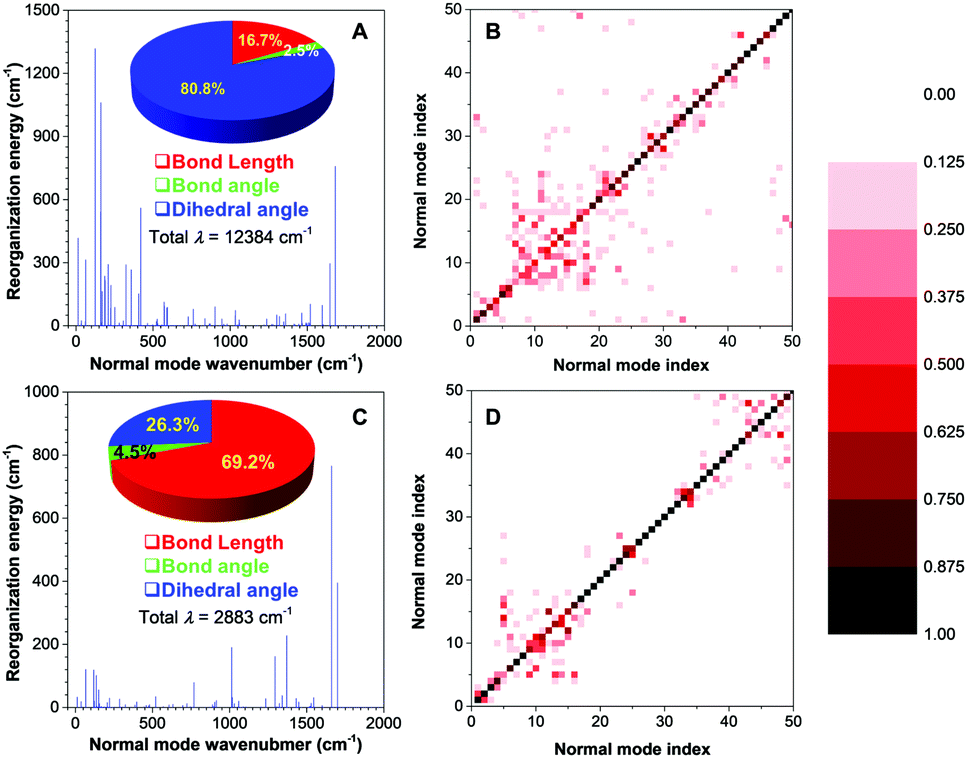 | ||
| Fig. 4 Quantum mechanics simulation on 2,4,5-TMe-DPE and 2,4,6-TMe-DPE. Plots of calculated reorganization energy (λ) versus the normal mode wavenumber of (A) 2,4,6-TMe-DPE and (C) 2,4,5-TMe-DPE in the gas phase calculated by the TD-DFT, B3LYP/6-311G** and Gaussian 09 program. Inset: Contribution of bond length, bond angle and dihedral angle to the total reorganization energy. Contour maps of the Duschinsky rotation matrix for the lowest 50 modes in (B) 2,4,6-TMe-DPE and (D) 2,4,5-TMe-DPE in the gas phase. | ||
Fig. 5A and B show the conformational change from S0 to S1. The conformational overlap in 2,4,5-TMe-DPE is obviously larger than that in 2,4,6-TMe-DPE. Detailed information can be found in Fig. 5C and D. From S0 to S1, the changes in twist angles α and α′ in 2,4,6-TMe-DPE were 62° and 22°, which were bigger than those of 2,4,5-TMe-DPE where changes in α and α′ were 18° and 26°, respectively. Meanwhile, a 20° change in double-bond torsion angle (β) was observed in 2,4,6-TMe-DPE, but the double-bond in 2,4,5-TMe-DPE remained planar even in the excited state. The excited-state double-bond length (γ) of 2,4,6-TMe-DPE was longer than that of 2,4,5-TMe-DPE. Three-dimensional potential energy surfaces (3D-PES) of 2,4,6-TMe-DPE and 2,4,5-TMe-DPE with regard to the rotation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and the phenyl rings were calculated and are plotted in Fig. 5E and F, respectively. For each point in the surface, the torsion angles α, α′ and β were fixed and then excited-state optimization was performed to acquire the S1 and S0 energies. The 3D-PES was only plotted in the range of β = 180° to 130° because of non-convergence at β < 130°. As shown in Fig. 5E, the most stable ground-state structure of 2,4,6-TMe-DPE was located at point a. After absorbing a photon, the molecule was immediately excited to point b which became an unstable excited-state structure. Then, the excited-state conformation b relaxed to its minimum energy point c through vibrational relaxation. From point b to c, both phenyl-ring twisting and double-bond torsion occurred. However, in 2,4,5-TMe-DPE (Fig. 5F), only phenyl-ring twisting was observed but without obvious ESDBT. In conclusion, we found that the twisted 2,4,6-TMe-DPE showed a large reorganization energy and most of its excited energy was released through nonradiative decay. In contrast, the planar 2,4,5-TMe-DPE had a rigid excited-state structure in which most of the nonradiative-decay channel was blocked. The calculation results were consistent with our experimental data that 2,4,5-TMe-DPE showed a high ΦF,solution = 13.4% but a negligible ΦF,solution of 0.6% for twisted 2,4,6-TMe-DPE. Further calculation revealed that ESDBT in 2,4,6-TMe-DPE contributed a lot to the predominant nonradiative decay. However, undetectable ESDBT was observed in planar 2,4,5-TMe-DPE, which resulted in a diminished nonradiative decay.
C double bond and the phenyl rings were calculated and are plotted in Fig. 5E and F, respectively. For each point in the surface, the torsion angles α, α′ and β were fixed and then excited-state optimization was performed to acquire the S1 and S0 energies. The 3D-PES was only plotted in the range of β = 180° to 130° because of non-convergence at β < 130°. As shown in Fig. 5E, the most stable ground-state structure of 2,4,6-TMe-DPE was located at point a. After absorbing a photon, the molecule was immediately excited to point b which became an unstable excited-state structure. Then, the excited-state conformation b relaxed to its minimum energy point c through vibrational relaxation. From point b to c, both phenyl-ring twisting and double-bond torsion occurred. However, in 2,4,5-TMe-DPE (Fig. 5F), only phenyl-ring twisting was observed but without obvious ESDBT. In conclusion, we found that the twisted 2,4,6-TMe-DPE showed a large reorganization energy and most of its excited energy was released through nonradiative decay. In contrast, the planar 2,4,5-TMe-DPE had a rigid excited-state structure in which most of the nonradiative-decay channel was blocked. The calculation results were consistent with our experimental data that 2,4,5-TMe-DPE showed a high ΦF,solution = 13.4% but a negligible ΦF,solution of 0.6% for twisted 2,4,6-TMe-DPE. Further calculation revealed that ESDBT in 2,4,6-TMe-DPE contributed a lot to the predominant nonradiative decay. However, undetectable ESDBT was observed in planar 2,4,5-TMe-DPE, which resulted in a diminished nonradiative decay.
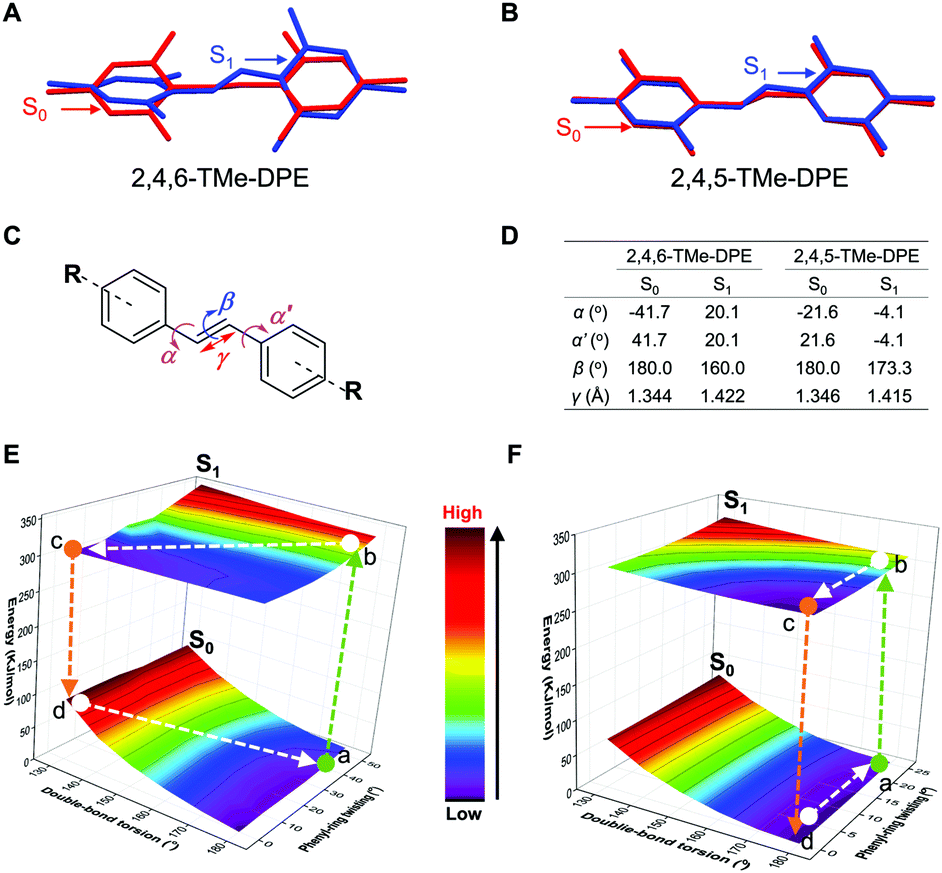 | ||
| Fig. 5 Quantum mechanics simulation on 2,4,5-TMe-DPE and 2,4,6-TMe-DPE. Simulated structures of (A) 2,4,6-TMe-DPE and (B) 2,4,5-TMe-DPE at the ground (S0) and first excited (S1) states in the gas phase. (C) Definition of the dihedral angle (α, α′), torsion angle (β) and bond length (γ). (D) Conformational change from S0 to S1. Three-dimensional potential energy surfaces of (E) 2,4,6-TMe-DPE and (F) 2,4,5-TMe-DPE in S0 and S1versus the double-bond torsion and phenyl-ring twisting. Calculated by the TD-DFT, B3LYP/6-311G** and Gaussian 09 program. | ||
To prove whether our proposed mechanistic picture is correct or not, another three pairs of systems were synthesized and characterized. Firstly, methyl groups in the 4-position were removed and 2,6-DMe-DPE and 2,5-DMe-DPE were obtained (Fig. S30 and S31, ESI†). These two molecules showed almost the same photophysical properties as their counterparts 2,4,6-TMe-DPE and 2,4,5-TMe-DPE. For example, the twisted 2,6-DMe-DPE was a typical AIEgen with ΦF,solution = 0.6%, but its isomeride 2,5-DMe-DPE exhibited strong emission in solution with ΦF,solution = 8.6%, which indicated that the para-position modification had little effect on their photophysical properties. In order to remove the effect of ESDBT, 2,4,6-TMe-TPh and 2,4,5-TMe-TPh were designed and the middle double bond was replaced by a benzene ring (Fig. S32, ESI†). The optimized dihedral angles between the core and sideward trimethylbenzene were 90° and 50° in 2,4,6-TMe-TPh and 2,4,5-TMe-TPh, respectively. As expected, the twisted 2,4,6-TMe-TPh already showed strong emission in solution (Fig. 6A) and its ΦF,solution increased to 1.8%. Fig. 6C suggests that 2,4,6-TMe-TPh exhibited an aggregation-enhanced emission effect but was not a typical AIEgen. Meanwhile, planar 2,4,5-TMe-TPh exhibited a weak ACQ effect which was similar to 2,4,5-TMe-DPE and 2,5-DMe-DPE (Fig. 6B and C). In a comparison between 2,4,6-TMe-TPh and 2,4,6-TMe-DPE, both of them were twisted structures and 2,4,6-TMe-TPh was even more twisted, but the stronger emission of 2,4,6-TMe-TPh in solution proved that ESDBT was an essential factor to facilitate nonradiative decay. Another example is the comparison between tetraphenylethylene (TPE) and tetraphenylbenzene (TPB). TPE is a widely used AIEgen which shows undetectable ΦF,solution.42,43 As shown in Fig. 6D, there was almost no emission when fw < 80% but a strong emission was induced with a further increase in molecular aggregation. Fig. 6F indicates that the emission intensity has been increased more than 500 times from fw = 0 to 90%. However, when its middle double bond was replaced by benzene, the resultant TPB already showed strong emission in pure THF solution with ΦF,solution = 1.5% (Fig. 6E and Fig. S33, ESI†). The photophysical behavior of TPB in THF/water mixtures was quite similar to the performance of 2,4,6-TMe-TPh (Fig. 6F).44
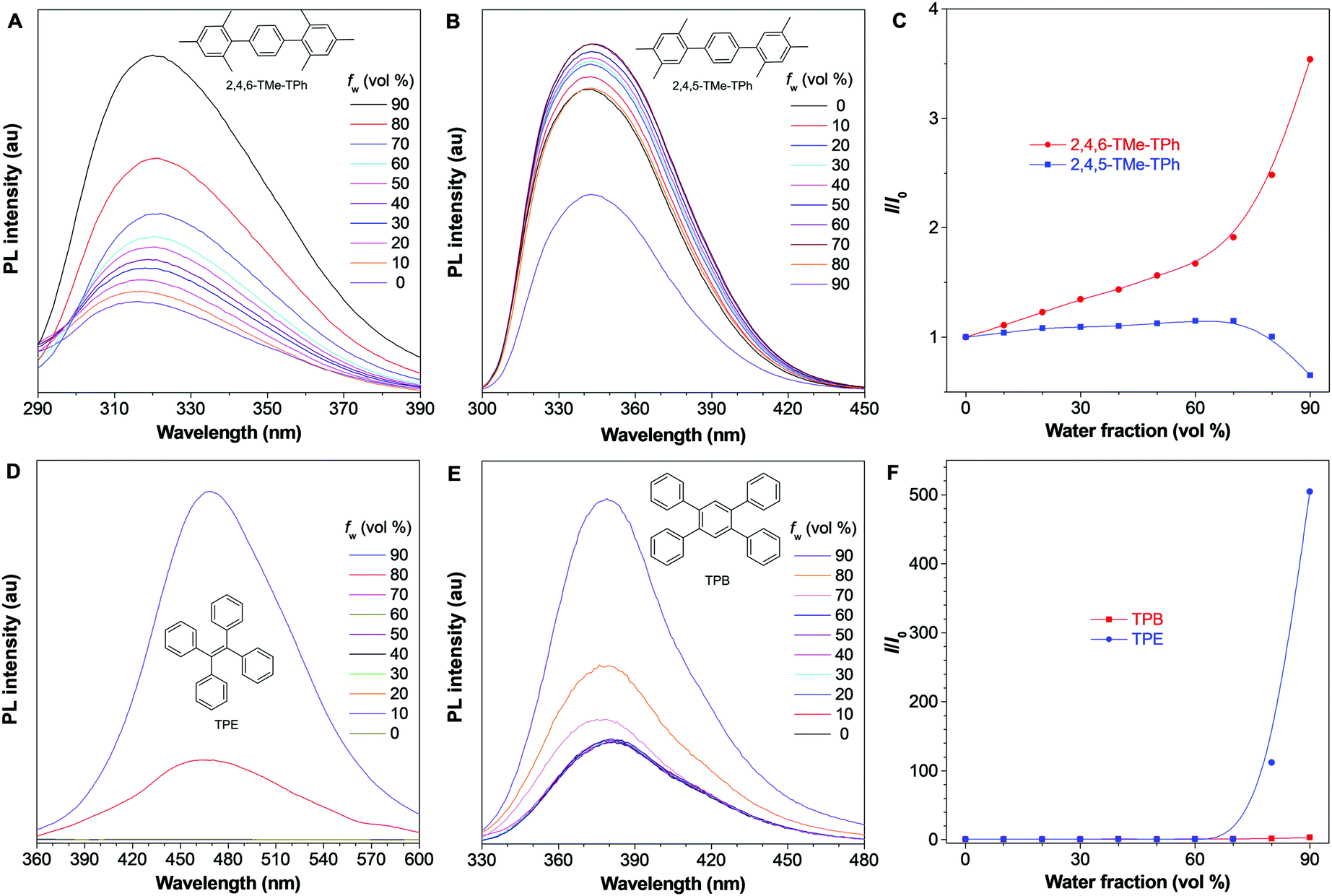 | ||
| Fig. 6 Photophysical studies on other fluorophores. PL spectra of (A) 2,4,6-TMe-TPh, (B) 2,4,5-TMe-TPh, (D) TPE and (E) TPB in THF/water mixtures with different water fractions (fw). Plots of relative PL intensity (I/I0) of (C) 2,4,6-TMe-TPh, 2,4,5-TMe-TPh and (F) TPE, TPB versus the composition of their THF/water mixture. Concentration = 10 μM, λex = 260 nm @ 2,4,6-TMe-TPh, 280 nm @ 2,4,5-TMe-TPh, 340 nm @ TPE and 275 nm @ TPB. I0 = PL intensity at fw = 0%. | ||
Conclusions
In summary, we have delved into the photophysical properties of two stilbene derivatives di-o-methyl substituted 2,4,6-TMe-DPE and mono-o-methyl substituted 2,4,5-TMe-DPE. Experimental results suggested that the twisted 2,4,6-TMe-DPE was nonluminescent in solution with a 0.6% ΦF,solution. In contrary, its isomeride 2,4,5-TMe-DPE showed a planar structure and a high ΦF,solution of 13.4% was observed. Time-resolved spectroscopy measurements on these two molecules indicated that the twisted 2,4,6-TMe-DPE possessed a faster and easier excited-state molecular motion than planar 2,4,5-TMe-DPE. Furthermore, reorganization energy and Duschinsky rotation matrix calculation also proved that the nonradiative decay was predominant in 2,4,6-TMe-DPE, but this channel was blocked in 2,4,5-TMe-DPE, which was consistent with the experimental data. Meanwhile, ESDBT was revealed to play an important role in the predominant nonradiative decay. Thus, we designed another three pairs of systems whose photophysical properties also proved the crucial role of ESDBT. These results draw a clear photophysical picture for this kind of organic luminophore, which makes their luminescent behavior predictable and controllable.Author contributions
H. Z. and B. T. designed the experiments. H. Z. synthesized all the compounds and measured all the PL, UV, QY, NMR, MS spectra. H. Z., J. L., Y. N., A. Q. and Q. P. carried out the theoretical calculation and result analysis. L. D., C. M., K. S. W. and D. L. P. did the time-resolved spectroscopy measurements. H. Z., N. L. C. L. and B. T. discussed and proposed the mechanistic picture. J. S., H. H. Y. S. and I. D. W. performed XRD measurement and crystal structure analysis. H. Z., J. W. Y. L. and R. T. K. K. revised the manuscript. H. Z. and B. T. wrote the manuscript with comments from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Science Foundation of China (21788102), the Research Grants Council of Hong Kong (16308016, N-HKUST604/14, C2014-15G, C6009-17G and A-HKUST 605/16), the University Grants Committee of Hong Kong (AoE/P-03/08 and AoE/P-02/12), the Innovation and Technology Commission (ITC-CNERC14SC01 and ITS/254/17) and the Science and Technology Plan of Shenzhen (JCYJ20160229205601482 and JCY20170818113602462). The University of Hong Kong Development Fund 2013-2014 project “New Ultrafast Spectroscopy Experiments for Shared Facilities”.References
- B. Valeur and M. N. Berberan-Santos, A Brief History of Fluorescence and Phosphorescence before the Emergence of Quantum Theory, J. Chem. Educ., 2011, 88, 731–738 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, 2010 Search PubMed.
- G. G. Stokes, On the Change of Refrangibility of Light, Philos. Trans. R. Soc. London, 1852, 142, 463–562 CrossRef.
- G. G. Stokes, On the Change of Refrangibility of Light. No. II, Philos. Trans. R. Soc. London, 1853, 143, 385–396 CrossRef.
- T. Förster and K. Kasper, Ein Konzentrationsumschlag der Fluoreszenz, Z. Phys. Chem., 1954, 1, 275–277 CrossRef.
- J. B. Birks, C. L. Braga and M. D. Lumb, ‘Excimer’ Fluorescence. VI. Benzene, Toluene, p-Xylene and Mesitylene, Philos. Trans. R. Soc., A, 1965, 283, 83–99 Search PubMed.
- S. T. Henderson, The development of luminescence since the 1938 Oxford conference, Br. J. Appl. Phys., 1955, 6, 1–3 CrossRef.
- E. N. Harvey, A history of luminescence from the earliest times until 1900, American Philosophical Society, Philadelphia, 1957 Search PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole, Chem. Commun., 2001, 1740–1741 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar!, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Synthesis, light emission, nanoaggregation, and restricted intramolecular rotation of 1,1-substituted 2,3,4,5-tetraphenylsiloles, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Control of aggregation-induced emission versus fluorescence aggregation-caused quenching by bond existence at a single site in boron pyridinoiminate complexes, Mater. Chem. Front., 2017, 1, 1573–1579 RSC.
- X. Ma, R. Sun, J. Cheng, J. Liu, F. Gou, H. Xiang and X. Zhou, Fluorescence Aggregation-Caused Quenching versus Aggregation-Induced Emission: A Visual Teaching Technology for Undergraduate Chemistry Students, J. Chem. Educ., 2016, 93, 345–350 CrossRef CAS.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, AIE luminogens: emission brightened by aggregation, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- M. Dommett, M. Rivera and R. Crespo-Otero, How Inter- and Intramolecular Processes Dictate Aggregation-Induced Emission in Crystals Undergoing Excited-State Proton Transfer, J. Phys. Chem. Lett., 2017, 8, 6148–6153 CrossRef CAS PubMed.
- Y. J. Gao, X. P. Chang, X. Y. Liu, Q. S. Li, G. Cui and W. Thiel, Excited-State Decay Paths in Tetraphenylethene Derivatives, J. Phys. Chem. A, 2017, 121, 2572–2579 CrossRef CAS PubMed.
- A. Prlj, N. Doslic and C. Corminboeuf, How does tetraphenylethylene relax from its excited states?, Phys. Chem. Chem. Phys., 2016, 18, 11606–11609 RSC.
- J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Locking the phenyl rings of tetraphenylethene step by step: understanding the mechanism of aggregation-induced emission, Chem. Commun., 2012, 48, 10675–10677 RSC.
- N. W. Tseng, J. Liu, J. C. Y. Ng, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Deciphering mechanism of aggregation-induced emission (AIE): Is E–Z isomerisation involved in an AIE process?, Chem. Sci., 2012, 3, 493–497 RSC.
- Q. Li and L. Blancafort, A conical intersection model to explain aggregation induced emission in diphenyl dibenzofulvene, Chem. Commun., 2013, 49, 5966–5968 RSC.
- K. Keshav, M. K. Kumawat, R. Srivastava and M. Ravikanth, Benzothiazoles-substituted tetraphenylethylenes: synthesis, structure, aggregation-induced emission and biological studies, Mater. Chem. Front., 2017, 1, 1207–1216 RSC.
- S. Liu, X. Zhou, H. Zhang, H. Ou, J. W. Y. Lam, Y. Liu, L. Shi, D. Ding and B. Z. Tang, Molecular Motion in Aggregates: Manipulating TICT for Boosting Photothermal Theranostics, J. Am. Chem. Soc., 2019, 141, 5359–5368 CrossRef CAS PubMed.
- J. Kim and T. M. Swager, Control of conformational and interpolymer effects in conjugated polymers, Nature, 2001, 411, 1030–1034 CrossRef CAS PubMed.
- H. Zhang, H. Li, J. Wang, J. Sun, A. Qin and B. Z. Tang, Axial chiral aggregation-induced emission luminogens with aggregation-annihilated circular dichroism effect, J. Mater. Chem. C, 2015, 3, 5162–5166 RSC.
- G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. Zhang and B. Z. Tang, Conjugation-Induced Rigidity in Twisting Molecules: Filling the Gap Between Aggregation-Caused Quenching and Aggregation-Induced Emission, Adv. Mater., 2015, 27, 4496–4501 CrossRef CAS PubMed.
- A. Vysniauskas, M. Balaz, H. L. Anderson and M. K. Kuimova, Dual mode quantitative imaging of microscopic viscosity using a conjugated porphyrin dimer, Phys. Chem. Chem. Phys., 2015, 17, 7548–7554 RSC.
- F. Wurthner, T. E. Kaiser and C. R. Saha-Moller, J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- W. J. Feast, P. W. Lövenich, H. Puschmann and C. Taliani, Synthesis and structure of 4,4′-bis(2,3,4,5,6-pentafluorostyryl)stilbene, a self-assembling J aggregate based on aryl–fluoroaryl interactions, Chem. Commun., 2001, 505–506 RSC.
- R. Hoffmann, Interaction of Orbitals through Space and through Bonds, Acc. Chem. Res., 1971, 4, 1–9 CrossRef CAS.
- H. Zhang, X. Zheng, N. Xie, Z. He, J. Liu, Ne. L. C. Leung, Y. Niu, X. Huang, K. S. Wong, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams, A. Qin, J. W. Y. Lam and B. Z. Tang, Why Do Simple Molecules with “Isolated” Phenyl Rings Emit Visible Light?, J. Am. Chem. Soc., 2017, 139, 16264–16272 CrossRef CAS PubMed.
- J. Sturala, M. K. Etherington, A. N. Bismillah, H. F. Higginbotham, W. Trewby, J. A. Aguilar, E. H. C. Bromley, A.-J. Avestro, A. P. Monkman and P. R. McGonigal, Excited-State Aromatic Interactions in the Aggregation-Induced Emission of Molecular Rotors, J. Am. Chem. Soc., 2017, 139, 17882–17889 CrossRef CAS PubMed.
- N. C. Greenham, I. D. W. Samuel, G. R. Hayes, R. T. Phillips, Y. A. R. R. Kessener, S. C. Moratti, A. B. Holmes and R. H. Friend, Measurement of Absolute Photoluminescence Quantum Efficiencies in Conjugated Polymers, Chem. Phys. Lett., 1995, 241, 89–96 CrossRef CAS.
- Y. Cai, L. Du, K. Samedov, X. Gu, F. Qi, H. H. Y. Sung, B. O. Patrick, Z. Yan, X. Jiang, H. Zhang, J. W. Y. Lam, I. D. Williams, D. L. Phillips, A. Qin and B. Z. Tang, Deciphering the working mechanism of aggregation-induced emission of tetraphenylethylene derivatives by ultrafast spectroscopy, Chem. Sci., 2018, 9, 4662–4670 RSC.
- S. A. Kovalenko, R. Schanz, H. Hennig and N. P. Ernsting, Cooling dynamics of an optically excited molecular probe in solution from femtosecond broadband transient absorption spectroscopy, J. Chem. Phys., 2001, 115, 3256–3273 CrossRef CAS.
- R. Berera, R. Grondelle and J. T. M. Kennis, Ultrafast transient absorption spectroscopy: principles and application to photosynthetic systems, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- Y. Ishibashi, M. Arinishi, T. Katayama, H. Miyasaka and T. Asahi, Femtosecond excited-state dynamics of fullerene-C60 nanoparticles in water, Phys. Chem. Chem. Phys., 2018, 20, 958–966 RSC.
- S. Park, A. L. Fischer, Z. Li, R. Bassi, K. K. Niyogi and G. R. Fleming, Snapshot Transient Absorption Spectroscopy of Carotenoid Radical Cations in High-Light-Acclimating Thylakoid Membranes, J. Phys. Chem. Lett., 2017, 8, 5548–5554 CrossRef CAS PubMed.
- Z. G. Shuai and Q. Peng, Organic light-emitting diodes: theoretical understanding of highly efficient materials and development of computational methodology, Natl. Sci. Rev., 2017, 4, 224–239 CAS.
- J. K. Liu, L. X. Pan, Q. Peng and A. J. Qin, Tetraphenylpyrimidine-Based AIEgens: Facile Preparation, Theoretical Investigation and Practical Application, Molecules, 2017, 22, 1679 CrossRef PubMed.
- T. Zhang, Q. Peng, C. Quan, H. Nie, Y. Niu, Y. Xie, Z. Zhao, B. Z. Tang and Z. Shuai, Using the isotope effect to probe an aggregation induced emission mechanism: theoretical prediction and experimental validation, Chem. Sci., 2016, 7, 5573–5580 RSC.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, Excited state radiationless decay process with Duschinsky rotation effect: formalism and implementation, J. Chem. Phys., 2007, 126, 114302 CrossRef PubMed.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li and B. Z. Tang, Aggregation-induced emissions of tetraphenylethene derivatives and their utilities as chemical vapor sensors and in organic light-emitting diodes, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- H. Zhang, X. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Tang, J. W. Y. Lam, A. Qin and B. Z. Tang, In situ monitoring of molecular aggregation using circular dichroism, Nat. Commun., 2018, 9, 4961 CrossRef PubMed.
- L. Li, M. Chen, H. Zhang, H. Nie, J. Z. Sun, A. Qin and B. Z. Tang, Influence of the number and substitution position of phenyl groups on the aggregation-enhanced emission of benzene-cored luminogens, Chem. Commun., 2015, 51, 4830–4833 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1481602, 1522067, 1564140 and 1585455. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00156e |
| ‡ These two authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |