
A fluorescence and photoactivity dual-activatable prodrug with self-synergistic magnification of the anticancer effect†
Jun
Li‡
a,
Xiang
Ni‡
a,
Jingtian
Zhang
a,
Yong
Liang
*b,
Zhiyuan
Gao
a,
Xiaoyan
Zhang
a,
Donghui
Zheng
*b and
Dan
Ding
 *ac
*ac
aKey Laboratory of Bioactive Materials Ministry of Education, College of Life Sciences, and State Key Laboratory of Medicinal Chemical Biology, Nankai University, Tianjin 300071, China. E-mail: dingd@nankai.edu.cn
bDepartment of Nephrology, Clinical Laboratory, Huai'an Hospital Affiliated to Xuzhou Medical University and Huai'an Second Hospital, Huai'an 223002, China. E-mail: harcyyly@163.com; zddwjj@126.com
cState Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
First published on 24th April 2019
Abstract
Reducing the chemo-resistance of cancer cells or making cancer cells sensitive to chemo-drugs plays a key role in boosting the efficacy of cancer chemotherapeutics in the clinic. Aiming for this, we develop a fluorescence and photoactivity dual-activatable prodrug (HCA–SS–HCPT) consisting of an aggregation-induced emission luminogen (AIEgen) named HCA (4-dimethylamino-2′-hydroxychalcone), a disulfide bond linker (–SS–), and the anticancer chemo-drug hydroxycamptothecin (HCPT). HCA–SS–HCPT is then formulated into nanoparticles with an average size of about 126 nm, which are weakly emissive and generate negligible reactive oxygen species (ROS) in aqueous media due to the quenching effect of the conjugated HCPT. In the presence of glutathione, whose concentration inside cancer cells is >1000-fold higher than that in blood, the –SS– is cleaved and then intact HCPT and HCA are simultaneously released, leading to restoration of the fluorescence and ROS generation capacity of HCA. Both in vitro and in vivo studies demonstrate that the cleaved HCA can not only track the co-delivered HCPT, but also more importantly, serve as a non-toxic pro-oxidant by production of a small amount of ROS under light irradiation to make four kinds of cancer cells much more susceptible to HCPT through oxidation therapy, thus achieving significant synergistic enhancement of anticancer efficacy.
Introduction
The research toward addressing the challenge of cancer cell resistance to chemo-drugs has received considerable interest, as chemo-resistance is one of the most important hindrances for effective cancer chemotherapeutics in the clinic.1–3 For instance, hydroxycamptothecin (HCPT) is one of the main components of first-line chemotherapy regimens for patients with advanced or metastatic colorectal cancer, as recommended by the national comprehensive cancer network (NCCN) guideline for colorectal cancer. For patients who are resistant/insensitive to HCPT, there is no standard chemotherapy regimen recommended.4,5 Moreover, peritoneal metastasis is considered as a final stage of ovarian cancer and colorectal cancer, and HCPT is widely used in patients with peritoneal metastasis to control the growth of malignant ascites owing to its effectiveness to treat peritoneal metastasis.6,7 If the peritoneal metastatic ovarian/colorectal cancer cells are resistant/insensitive to HCPT, the patients' overall survival time would be significantly shortened. As a consequence, it is of great significance to reduce the chemo-resistance of cancer cells to HCPT.It has been well established that oxidation therapy is an efficient approach to abate cancer cell resistance or make cancer cells sensitive to chemo-drugs such as paclitaxel and HCPT.8–10 Oxidation therapy often requires pre-/co-administration of a pro-oxidant to enable cancer cells to generate an intracellular oxidative microenvironment for example to lead to increased intracellular endogenous reactive oxygen species (ROS), which plays a pivotal role in significant amplification of the killing effect of chemo-drugs against cancer cells that are resistant or insensitive to the chemo-drugs.11–13 However, the currently available pro-oxidants for oxidation therapy are usually cytotoxic to normal tissues/cells,8 which decidedly causes the risk of severe side effects, resulting in elevated suffering of patients. Thereby, alternative oxidation therapy strategies using safe and non-toxic pro-oxidants are urgently being pursued.
Photodynamic therapy (PDT) through light irradiation of a photosensitizer to produce ROS that are mainly singlet oxygen holds the advantages of in situ cancer cell killing as well as spatial–temporal controllability.14–17 This motivates us to investigate whether such spatial–temporal tunable ROS generation by “photosensitizer + light irradiation” can serve as an effective “pro-oxidant” to enhance the efficacy of chemo-drugs. As for photosensitizers, the ones with aggregation-induced emission (AIE) characteristics often possess a three-dimensional twisted molecular structure, and are particularly useful, since they conquer the concentration/aggregation-caused attenuation of ROS production suffered by conventional photosensitizers with a planar molecular structure.18–21 Besides efficient ROS generation in biological environments, the AIE photosensitizers also feature concurrent high fluorescence, excellent anti-photobleaching and anti-ROS stability, as well as low cytotoxicity and in vivo toxicity.22–28 The momentous advantages of AIE photosensitizers make them promising for sophisticated biomedical applications and potential clinical translation.
To the best of our knowledge, there are rather few studies on the utilization of photosensitizers as a non-toxic pro-oxidant for oxidation therapy, i.e., synergistically addressing the chemo-resistance or insensitivity issue of cancer cells to chemo-drugs.19,29 In this contribution, we combine the advantages of AIE photosensitizers, oxidation therapy, and tumor microenvironment-responsive systems by design and synthesis of an AIE luminogen (AIEgen)-based, fluorescence and photoactivity dual-activatable HCPT prodrug (HCA–SS–HCPT, Scheme 1). The compound HCA–SS–HCPT consists of an AIEgen 4-dimethylamino-2′-hydroxychalcone (HCA),30 a disulfide bond linker, and HCPT. Formulation of HCA–SS–HCPT using an amphiphilic co-polymer as the encapsulation matrix affords nanoparticles (NPs) that transfer the hydrophobic HCA–SS–HCPT into water and allow for suitable in vivo application. The HCA–SS–HCPT NPs are almost non-fluorescent and hardly generate ROS under light irradiation in aqueous environments. After thiol (e.g., glutathione (GSH), in high concentrations within many cancer cells)31–34 responsiveness with cleavage of the disulfide bond, followed by release of intact HCPT and HCA (Scheme 2), the fluorescence and ROS generation ability of the AIEgen are restored, which can not only track the co-delivered HCPT, but also more importantly, serve as a non-toxic pro-oxidant by producing ROS to synergistically amplify the anticancer effect of HCPT by virtue of oxidation therapy. Noteworthily, by optimizing the light irradiation condition, the thiol-activated HCA can only generate a small amount of ROS, which is not high enough to kill cancer cells, but just provides an intracellular oxidative environment for oxidation therapy. The working principle and self-synergistic anticancer effect of HCA–SS–HCPT NPs are further demonstrated using cancer cells and tumor-bearing nude mice.
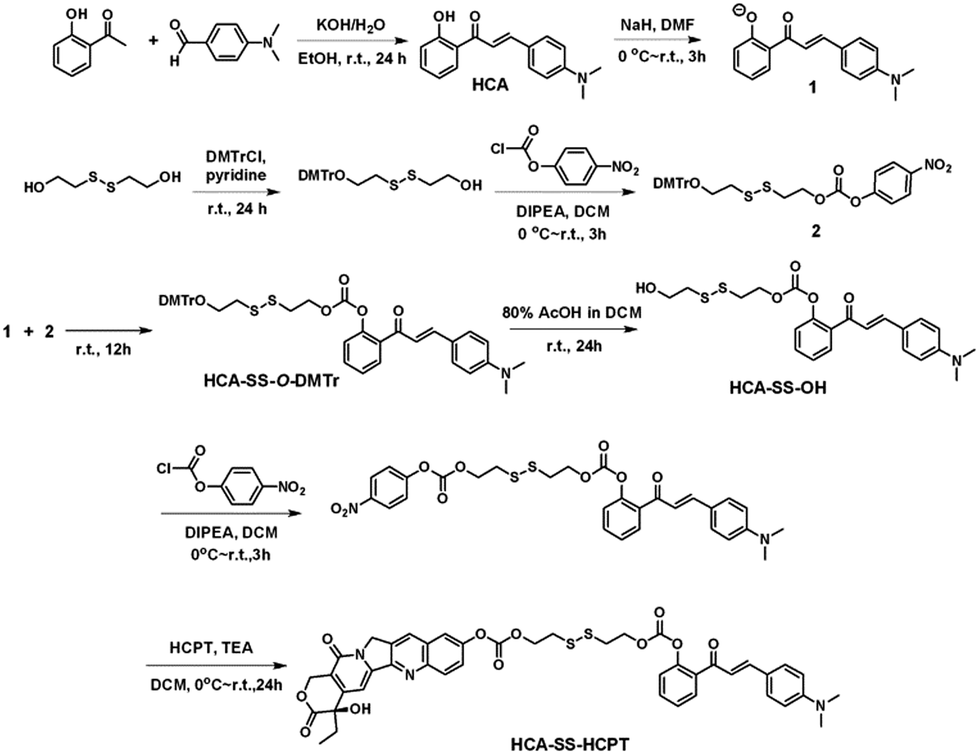 | ||
| Scheme 1 The synthetic route to HCA–SS–HCPT. | ||
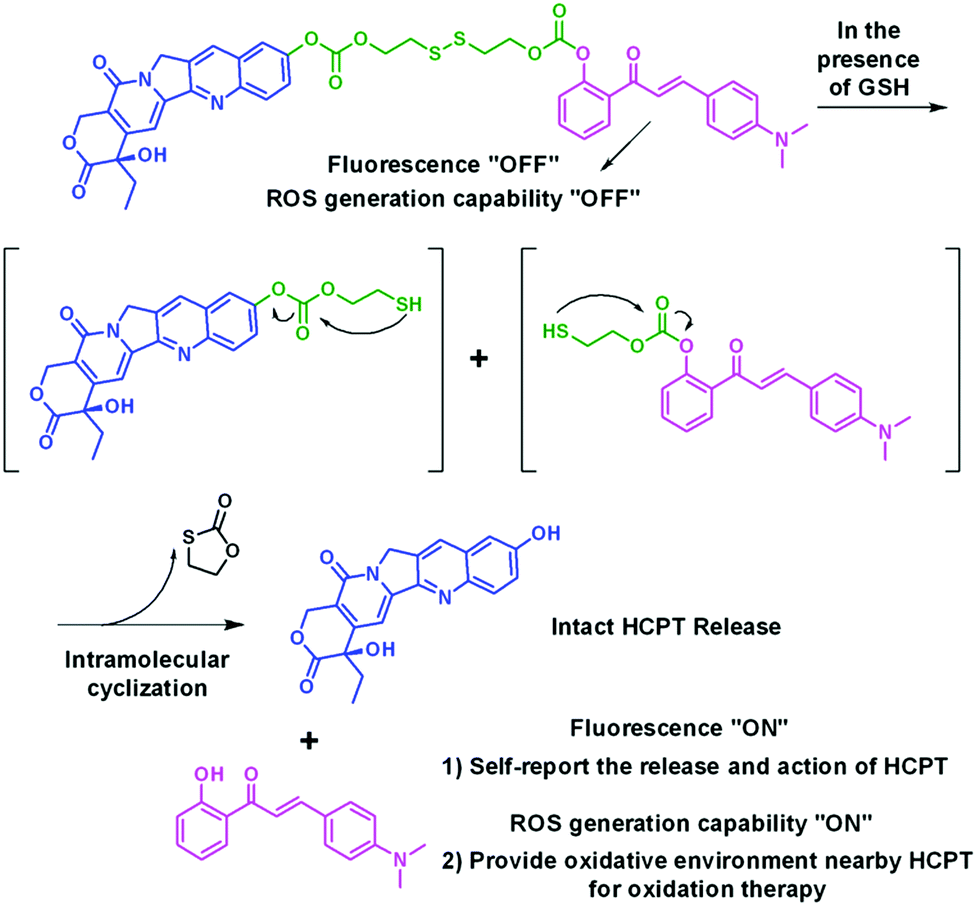 | ||
| Scheme 2 Design rationale and working principle of HCA–SS–HCPT. | ||
Results and discussion
The synthetic route to HCA–SS–HCPT is shown in Scheme 1. The compound HCA that has been demonstrated to be AIE-active was synthesized according to the literature.30 In this work, we first verify that HCA is also a good photosensitizer (Fig. S1, ESI†). HCA was reacted with the inorganic base NaH to form a negative ion, which was then added into the mono-O-dimethoxytrityl-2-hydroxyethyl disulfide solution to afford the intermediate HCA–SS–O-DMTr, followed by removal of the protective group O-DMTr to obtain the compound HCA–SS–OH. Subsequently, HCA–SS–OH was reacted with 4-nitrophenyl chloroformate to yield a reactive intermediate and HCPT was then added to afford the final product HCA–SS–HCPT. 1H NMR, 13C NMR and HRMS were used to characterize HCA–SS–OH and HCA–SS–HCPT, confirming their identity and purity (Fig. S2–S7, ESI†).The design and working principle of HCA–SS–HCPT are illustrated in Scheme 2. It is hypothesized that both the fluorescence and ROS generation capability of the AIEgen HCA can be quenched by the conjugation of HCPT, leading to HCA–SS–HCPT exisiting in both the “fluorescence OFF” and “ROS generation OFF” states. Besides, due to the chemical modification of a large moiety, HCPT loses its anticancer activity and HCA–SS–HCPT also serves as a prodrug. When HCA–SS–HCPT meets thiols, like GSH, which is a well-known cancer biomarker and is found at a much higher level within cancer cells than normal cells, the disulfide bond linker (–SS–) in HCA–SS–HCPT will become labile and self-immolative. After cleavage of the disulfide bond, the unstable residues are expected to undergo an intramolecular cyclization reaction,35–51 followed by release of intact HCPT and HCA. The intact HCPT will then exert its anticancer activity, and meanwhile, HCA restores its fluorescence and ROS generation capability, which hence not only self-reports the release and action of HCPT by the “turn-on” fluorescence, but more importantly, can provide an in situ oxidative environment near HCPT upon light irradiation, realizing an enhanced anticancer effect of HCPT against its resistant/insensitive cancer cells by oxidation therapy.
To test the hypothesis, a series of experiments were performed. First of all, to enable the hydrophobic HCA–SS–HCPT to access aqueous biological environments, a biocompatible co-polymer 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) was employed as the doping matrix to formulate HCA–SS–HCPT into NPs (named HCA–SS–HCPT NPs for short; Fig. 1A) via a nanoprecipitation approach. Dynamic light scattering (DLS) and transmission electron microscopy (TEM) data reveal that the HCA–SS–HCPT NPs possess a mean hydrodynamic diameter of around 126 nm as well as a nearly spherical morphology (Fig. 1B). The appropriate NP size and surface chemistry make the HCA–SS–HCPT NPs suitable for in vivo utilization.52 In addition, the NPs have good colloidal stability, with their average size negligibly changing within a week (Fig. S8, ESI†).
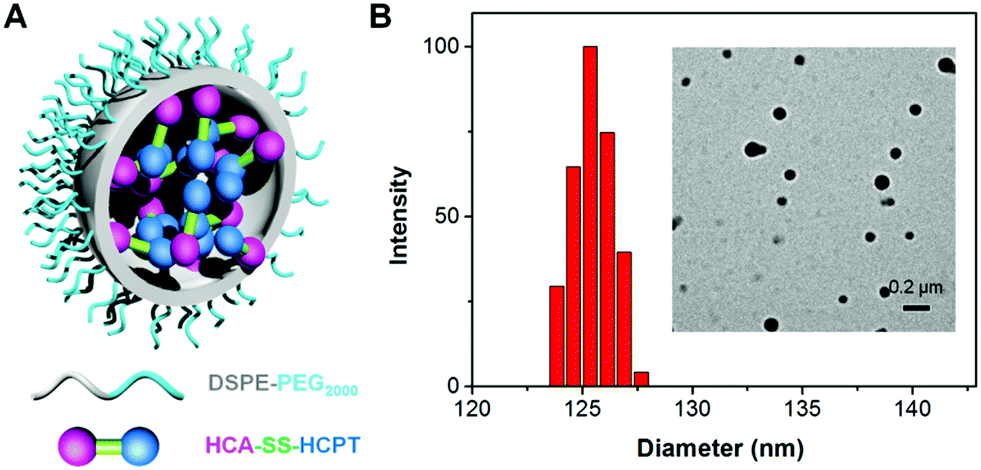 | ||
| Fig. 1 (A) Schematic illustration of HCA–SS–HCPT NPs with DSPE-PEG2000 as the doping matrix. (B) DLS profile (polydispersity (PDI): 0.15) and TEM image (inset) of HCA–SS–HCPT NPs. | ||
Subsequently, the optical properties of HCA–SS–HCPT NPs in the absence and presence of GSH are investigated in phosphate buffered saline (PBS). The HCA–SS–HCPT NPs in PBS buffer (pH 7.4) have an absorption maximum at 370 nm with a large shoulder centered at 425 nm (Fig. S9, ESI†), but are weakly emissive in aqueous media (Fig. 2A), because of the fluorescence quenching effect of the conjugated HCPT. After the addition of GSH, the disulfide bond of HCA–SS–HCPT between HCA and HCPT is cleaved, followed by release of intact HCA and HCPT. The “HCA–SS–HCPT NPs (10 μM based on HCA–SS–HCPT) + GSH (1 mM)” (co-incubation for 3 h at 37 °C) have a broad absorption peak centered at 412 nm (Fig. S9, ESI†). Moreover, as shown in Fig. 2A, the fluorescence intensity peaking at 560 nm is significantly enhanced by 20 fold after activation by GSH. Rather bright yellow fluorescence from the PBS solution of HCA–SS–HCPT NPs post incubation with GSH for 3 h at 37 °C can be seen under illumination by a 365 nm lamp (Fig. 2A). In addition, the HCA–SS–HCPT NP solution's fluorescence intensity centered at 560 nm gradually increases with increasing GSH concentration from 0 to 60 equiv. of the HCA–SS–HCPT molecule (Fig. 2B). It is also found that the pH value of the PBS solution of HCA–SS–HCPT NPs plays a key role in emission enhancement after reaction with GSH. As depicted in Fig. 2C, the fluorescence turn-on ratios under weak basic and neutral conditions are much higher than those under acidic conditions.
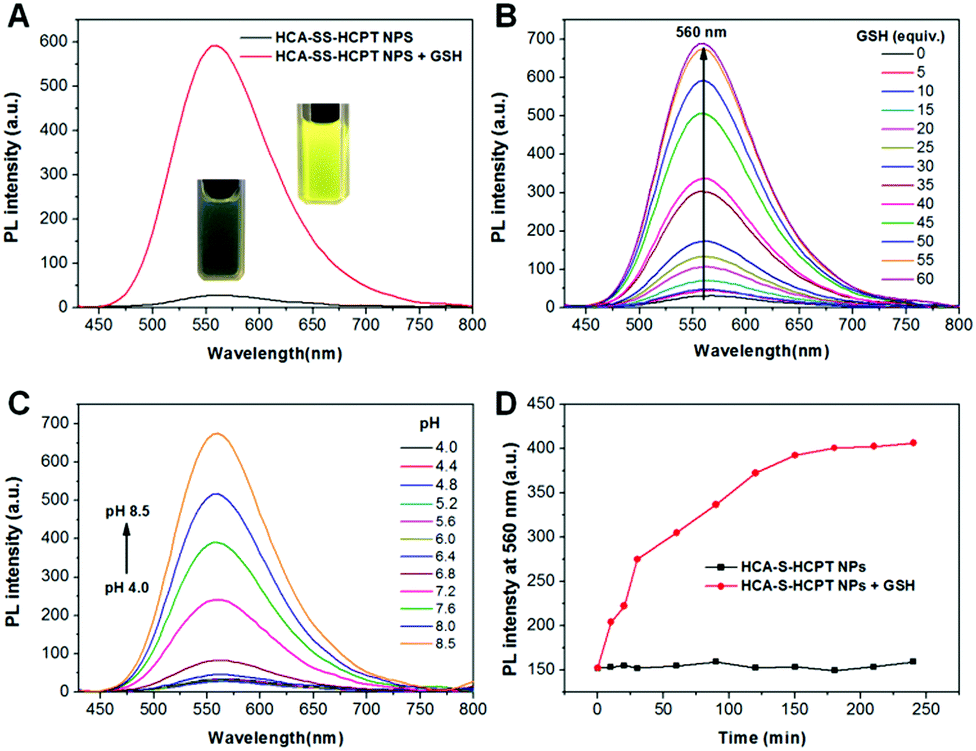 | ||
| Fig. 2 (A) Photoluminescence (PL) spectra of HCA–SS–HCPT NPs in PBS (pH = 7.4) recorded in the presence and absence of GSH (1 mM). Inset: Photographs of HCA–SS–HCPT NPs before and after GSH treatment taken under illumination with a 365 nm lamp. (B) Fluorescence changes of HCA–SS–HCPT NPs upon treatment with increasing concentrations of GSH (0–60 equiv. of HCA–SS–HCPT). (C) PL spectra of HCA–SS–HCPT NPs in the presence of GSH (1 mM) as a function of pH values of PBS buffers. (D) Time-dependent changes in fluorescence intensity at 560 nm of HCA–SS–HCPT NPs with and without the addition of GSH (1 mM) in PBS solution (pH = 7.4). For (A–D), excitation at 405 nm; [HCA–SS–HCPT NPs] = 10 μM based on the HCA–SS–HCPT molecule; GSH incubation/reaction time: 3 h; incubation temperature: 37 °C. | ||
The kinetics of fluorescence activation resulting from HCA–SS–HCPT cleavage by GSH was then studied. After addition of 1 mM GSH to the PBS solution of HCA–SS–HCPT NPs (10 μM based on HCA–SS–HCPT), the fluorescence intensity at 560 nm was monitored at 37 °C over time. As shown in Fig. 2D, the NP solution's fluorescence intensity gradually increases with time post addition of GSH, and reaches a plateau at around 3 h. The selectivity of HCA–SS–HCPT NPs toward thiols was studied. It is demonstrated that there is negligible fluorescence emission enhancement upon incubation with thiol-free amino acids and biologically relevant metal ions, including Na+, K+, Ca2+, Mg2+, Zn2+, Fe2+, Fe3+, and Cu2+, respectively (Fig. S10, ESI†). On the other hand, the amino acids with a thiol group such as cysteine (Cys) and homocysteine (Hcy) can light up the NP solution like GSH does (Fig. S10, ESI†). This result suggests that HCA–SS–HCPT NPs are selectively responsive to thiols.
We next assessed whether HCA–SS–HCPT NPs work in cancer cells by confocal laser scanning microscopy (CLSM). It has been reported that GSH is found at an extremely high level inside many cancer cells, which is >1000 fold that in blood.33,35,36 In our experiment, HeLa human cervical cancer cells were firstly employed. As displayed in Fig. 3A, under the used CLSM imaging condition, the cancer cell autofluorescence (without HCA–SS–HCPT NP incubation) is eliminated by optimizing the CLSM setting parameters.53 After co-incubating the HeLa cancer cells with HCA–SS–HCPT NPs (10 μM based on HCA–SS–HCPT) at 37 °C for 3 h, the subsequent CLSM imaging revealed that there is intense yellow fluorescence observed in the cytoplasm of HeLa cells (Fig. 3B), probably due to the reaction between HCA–SS–HCPT NPs and the intracellular GSH. To confirm this, the HeLa cells were pre-treated with a GSH inhibitor buthionine sulfoximine (BSO), followed by incubation with HCA–SS–HCPT NPs. As displayed in Fig. 3C, owing to the suppressed GSH level in the cells by BSO, the average fluorescence intensity in the BSO/HCA–SS–HCPT NP co-treated HeLa cells is far weaker than that in Fig. 3B. This result manifests the process of thiol-induced disulfide bond cleavage along with fluorescence turn-on. Furthermore, in several other kinds of cancer cells, such as PC-3 human prostate cancer cells, HepG2 human liver cancer cells and Caco-2 human colon cancer cells, we are also capable of observing the bright fluorescence switching on by CLSM imaging (Fig. 3D–F). The cellular imaging results together demonstrate that the HCA–SS–HCPT NPs are specifically activated by intracellular GSH in cancer cells, and the light-up fluorescence can report the release, location, and action of HCPT.
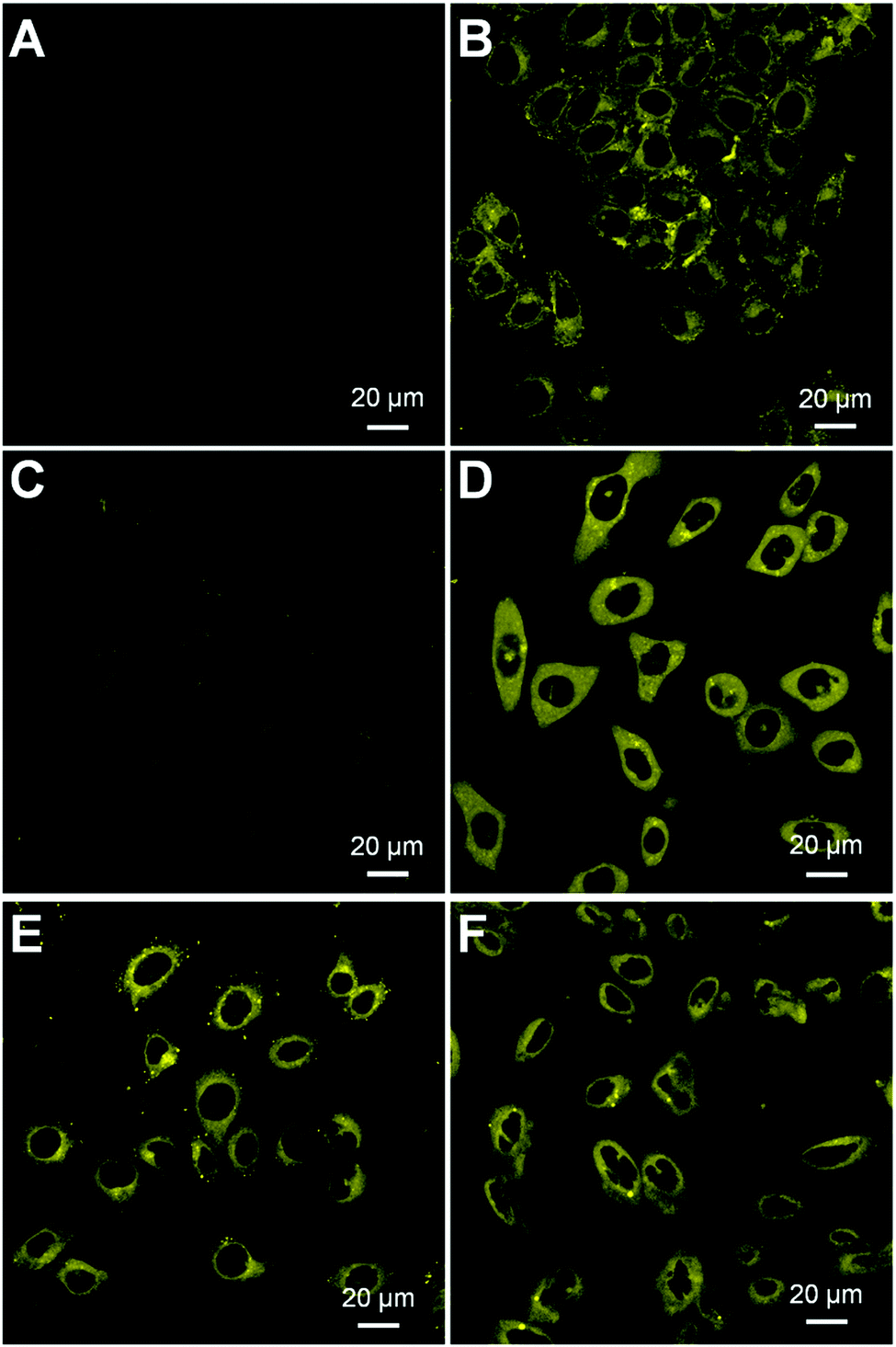 | ||
| Fig. 3 CLSM images of (A) HeLa cancer cells without incubation with HCA–SS–HCPT NPs, (B) HeLa cells incubated with HCA–SS–HCPT NPs at 37 °C for 3 h, and (C) HeLa cells pre-treated with BSO (100 μM) and further incubated with HCA–SS–HCPT NPs at 37 °C for 3 h. CLSM images of (D) PC-3, (E) HepG2 and (F) Caco-2 cancer cells incubated with HCA–SS–HCPT NPs at 37 °C for 3 h. For (B–F), [HCA–SS–HCPT NPs] = 10 μM based on HCA–SS–HCPT. | ||
The ROS generation capacities of HCA–SS–HCPT NPs in the absence and presence of GSH were evaluated using 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) as the ROS indicator, which is non-emissive but becomes fluorescent green post reaction with ROS to yield dichlorofluorescein (DCF).19 As shown in Fig. 4A, it is obvious that HCA–SS–HCPT NPs themselves in PBS are not able to produce ROS upon continuous white light (0.25 W cm−2) irradiation. Nevertheless, after HCA–SS–HCPT NPs are pre-activated by GSH, the fluorescence intensity from DCF at 530 nm shows large enhancement during the 3 min light exposure duration. Furthermore, the green fluorescence from DCF is significantly reduced when vitamin C is added to scavenge the produced ROS by HCA–SS–HCPT NPs with light irradiation. These results suggest that the ROS generation ability can recover along with fluorescence activation after reaction with GSH to yield free HCA.
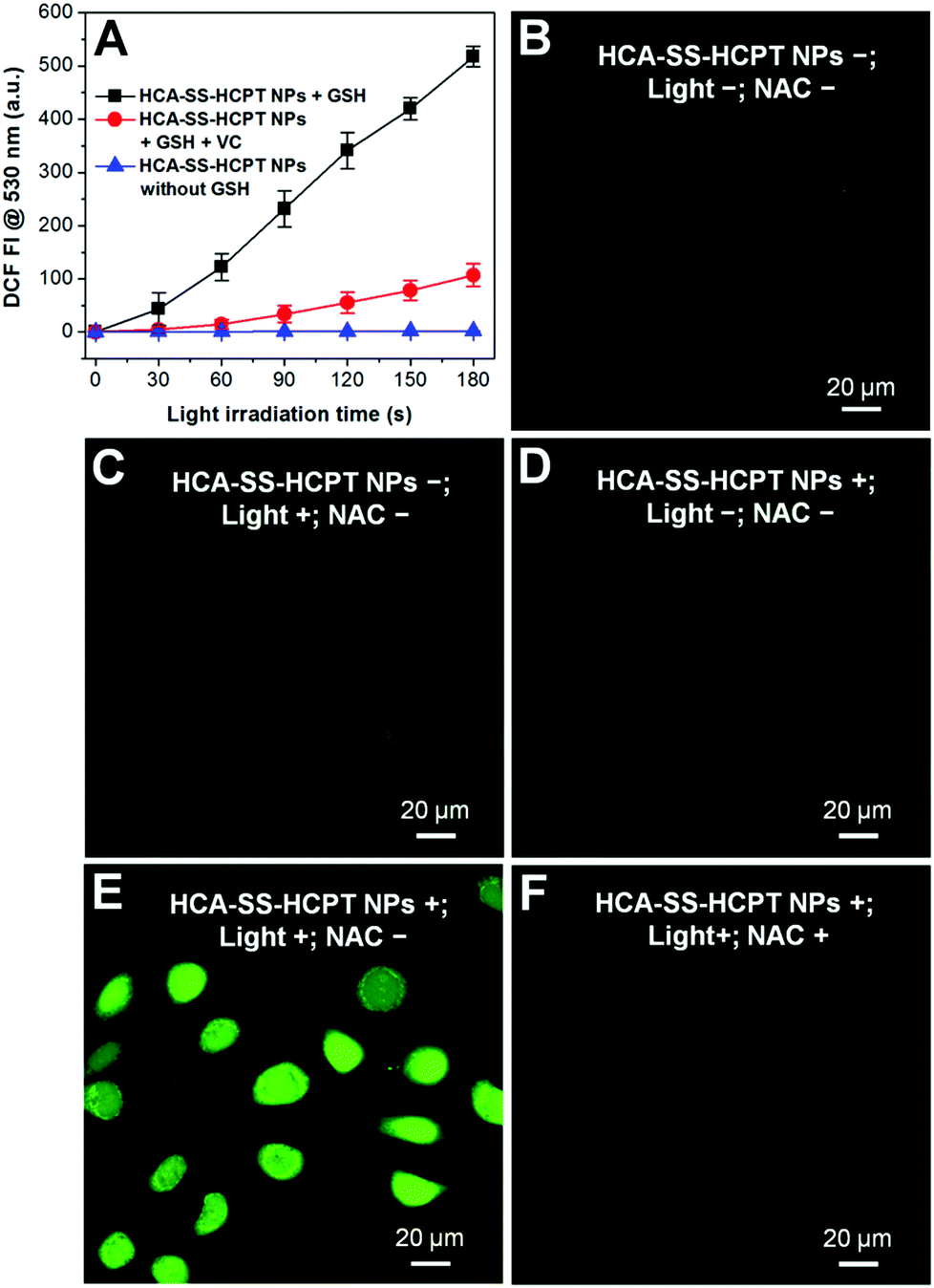 | ||
| Fig. 4 (A) Fluorescence intensity (FI) of DCF at 530 nm as a function of light irradiation time of HCA–SS–HCPT NPs in aqueous solution with and without the addition of GSH (or vitamin C (VC)). (B–F) CLSM images depict the intracellular ROS levels of HeLa cells receiving different treatments using DCFDA as the ROS indicator. The HeLa cells were incubated with HCA–SS–HCPT NPs (10 μM based on HCA–SS–HCPT) for 3 h. White light (400–700 nm) irradiation (0.25 W cm−2) was applied for 3 min. [NAC] = 1 mM. | ||
The reversibility of the ROS production capacity of HCA–SS–HCPT NPs was also assessed in cancer cells and DCFDA was employed as the ROS indicator as well. As a control, the antioxidant N-acetylcysteine (NAC) was used to treat the cancer cells before incubation with HCA–SS–HCPT NPs, since NAC can scavenge the ROS if generated. As shown in Fig. 4B–D, only weak green fluorescence from DCF can be seen for the HeLa cells themselves (NPs−; Light−; NAC−), HeLa cells with white light irradiation (NPs−; Light+; NAC−), and HCA–SS–HCPT NP-treated HeLa cells (NPs+; Light−; NAC−). In sharp contrast, as displayed in Fig. 4E, brilliant green emission of DCF is observed from the entire HeLa cells that are incubated with HCA–SS–HCPT NPs and then exposed to white light for 3 min (NPs+; Light+; NAC−). Such DCF fluorescence enhancement can be significantly inhibited in the presence of NAC (Fig. 4F). These results verify that the HCA–SS–HCPT NPs can generate ROS effectively within cancer cells by virtue of activation by intracellular thiols.
The anticancer activity of released HCPT and the probability of utilizing intracellular enhanced ROS to amplify the anticancer efficacy of released HCPT were verified in four cancer cell lines including HeLa, PC-3, HepG2, and Caco-2 cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. For better comparison, HCA–SS–OH (without HCPT conjugation) doped DSPE-PEG2000 NPs (HCA–SS–OH NPs for short) were prepared and used as a control. It is worth noting that “HCA–SS–OH NPs + GSH” has comparable ROS generation ability to “HCA–SS–HCPT NPs + GSH” in aqueous media.
After each cancer cell line was incubated with HCA–SS–OH NPs, white light irradiation (0.25 W cm−2, 3 min) was applied at 8 h, which was followed by MTT assay at 24 h in the dark. As shown in Fig. 5A–D, the 24 h cell viability at different NP concentrations based on HCA–SS–OH for all four kinds of cancer cells is >90% (HCA–SS–OH NPs+; Light+). The cell viability values are similar to the corresponding data of HCA–SS–OH NP-treated cells without exposure to white light (HCA–SS–OH NPs+; Light−). The result reveals that (1) the HCA–SS–OH NP itself is nearly non-toxic; and (2) under our experimental conditions (0.25 W cm−2 white light irradiation for 3 min), the ROS generated by the released HCA is at a low level, which can just provide an intracellular oxidative environment (as evidenced by the obvious green DCF fluorescence within cancer cells), but not kill cancer cells like PDT does.
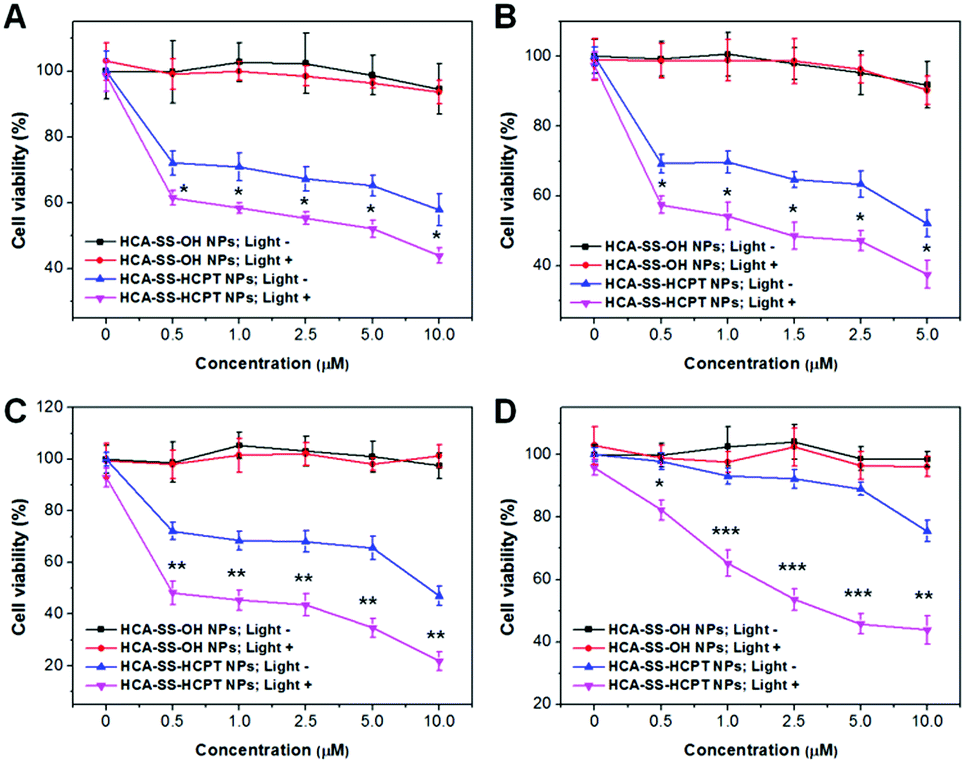 | ||
| Fig. 5 Cell viability of (A) HeLa, (B) PC-3, (C) HepG2 and (D) Caco-2 cancer cells upon treatment with various concentrations of HCA–SS–OH NPs and HCA–SS–HCPT NPs (based on HCA–SS–OH or HCA–SS–HCPT) in the dark or under white light irradiation (3 min, 0.25 W cm−2) at 8 h. The cells were incubated with the NPs for 24 h, followed by MTT assay. *, **, and *** represent P < 0.05, P < 0.01, and P < 0.001, respectively, in comparison between “HCA–SS–HCPT NPs; Light−” and “HCA–SS–HCPT NPs; Light+” groups. | ||
Then, we studied whether the intracellular oxidative environment provided by the GSH-activated HCA–SS–HCPT NPs could amplify the anticancer efficacy of the released HCPT. Four kinds of cancer cells were incubated with a variety of concentrations of HCA–SS–HCPT NPs (based on HCA–SS–HCPT). At 8 h post NP addition, the cells were irradiated by white light (0.25 W cm−2, 3 min). Alternatively, the NP-treated cells were kept in the dark. And then the MTT assay was carried out at 24 h in the dark. Of note, HCA–SS–HCPT NPs without light exposure (HCA–SS–HCPT NPs+; Light−) can suppress the cancer cells to a certain extent in a dose-dependent manner, demonstrating the efficient release of cytotoxic HCPT from the NPs by the action of intracellular thiols, which also manifests that HCA–SS–HCPT NPs serve as an effective thiol-responsive prodrug of HCPT. It is also noted that the four kinds of cancer cell lines we selected in this work are insensitive to HCPT. Even at the highest NP concentration (10 μM for HCPT), about 50% of HeLa, PC-3 and HepG2 cancer cells are still viable (Fig. 5A–C). Particularly, the Caco-2 cancer cells we used are quite resistant to HCPT, as evidenced by the 90% cell viability on treatment with 5 μM HCA–SS–HCPT NPs (Fig. 5D).
Encouragingly, if we endow the cancer cells in particular the Caco-2 cancer cells with an intracellular oxidative environment by exposure of the cancer cells to white light in order to enable the released HCA produce ROS, the cancer cells become far more sensitive to the released HCPT, leading to self-synergistic magnification of the anticancer effect. The differences in cell viability at each concentration for each cancer cell line between the treatments of HCA–SS–HCPT NPs with and without white light irradiation are statistically significant (Fig. 5). This result proves the hypothesis of our proposed self-synergistic oxidation therapy approach. As we use HCA–SS–OH NPs as a control to validate that the ROS generated by the cleaved HCA would not cause cell death, such synergistic enhancement of anticancer efficacy can be described as “0 + 1 > 1”.
We further estimated the theranostic performance of HCA–SS–HCPT NPs in vivo using a xenograft PC-3 tumor-bearing nude mouse model. After injecting HCA–SS–HCPT NPs (2.5 mg kg−1 based on HCPT) into the tumor-bearing mice via the tail vein, the tumors were collected for imaging using an in vivo Maestro small-animal fluorescence imaging instrument. It is found that the fluorescence from HCA–SS–HCPT NPs can be switched on in tumor tissues, whose intensity reaches the maximum at 5 h post-injection (Fig. S11, ESI†). Noteworthily, the yellow fluorescence signal from cleaved HCA is clearly observed in tumor slices collected at 5 h post-injection by CLSM, which is located close to the cell nuclei in the cytoplasm (Fig. 6A). These results reveal that HCA–SS–HCPT NPs reach the tumor tissues from the blood circulation by the enhanced permeability and retention (EPR) effect54–58 and the HCA emission can recover in the presence of intracellular GSH in tumors of live mice to indicate where the co-delivered HCPT unloads.
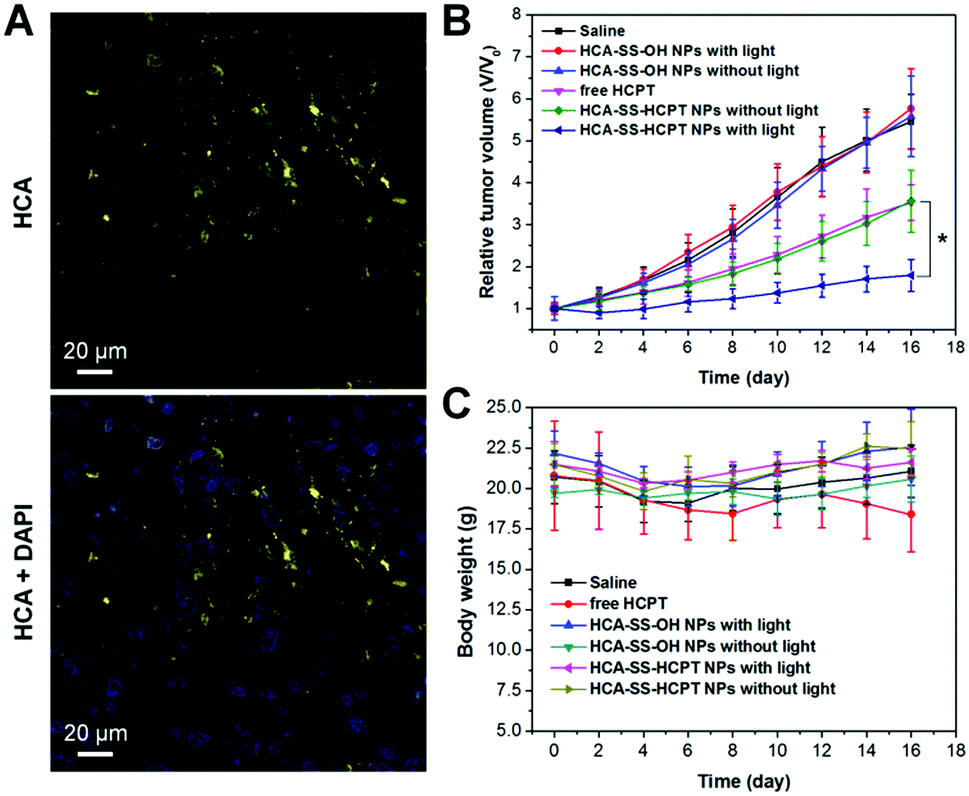 | ||
| Fig. 6 (A) Representative CLSM images of tumor slices from HCA–SS–HCPT NP-treated mice. The HCA–SS–HCPT NPs (2.5 mg kg−1 based on HCPT) were intravenously injected into tumor-bearing mice and at 5 h post-injection, the tumors were excised and sectioned. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). (B) Tumor growth curves of different treatments indicated. Data are presented as mean ± s.d. (n = 6 mice per group). * represents P < 0.05. (C) Body weight changes of mice in different groups. | ||
Next, we studied whether the self-synergetic HCPT oxidation antitumor therapy works in vivo. In this experiment, PC-3 tumor-bearing mice were divided into six groups randomly (each group includes 6 mice), which are named as follows: “Saline”, “free HCPT”, “HCA–SS–OH NPs without light”, “HCA–SS–OH NPs with light”, “HCA–SS–HCPT NPs without light”, and “HCA–SS–HCPT NPs with light”, respectively. For the treatments of “Saline”, “free HCPT”, “HCA–SS–OH NPs without light”, and “HCA–SS–HCPT NPs without light”, on day 0, saline, free HCPT (2.5 mg kg−1), HCA–SS–OH NPs (1.8 mg kg−1 based on HCA), and HCA–SS–HCPT NPs (2.5 mg kg−1 based on HCPT and 1.8 mg kg−1 based on HCA) were intravenously injected into the tumor-bearing mice, respectively, followed by tumor volume monitoring of each mouse over time. For the treatments of “HCA–SS–OH NPs with light” and “HCA–SS–HCPT NPs with light”, after intravenous administration of HCA–SS–OH NPs and HCA–SS–HCPT NPs, respectively, for 5 h, the tumor tissues of mice in these two cohorts were irradiated with white light (0.3 W cm−2) for 10 min. The antitumor effect of each treatment was then assessed during the following 16 day study duration.
As presented in Fig. 6B, the tumor growth of mice in “Saline”, “HCA–SS–OH NPs without light” and “HCA–SS–OH NPs with light” groups is fast and these treatments have negligible antitumor activity. This result indicates that the ROS generated by the cleaved HCA with light irradiation fail to kill cancer cells and suppress the tumor growth. On the other hand, free HCPT is efficacious in tumor volume inhibition to some extent; however, the average tumor volume on day 16 is still about 3.5-fold larger than that on day 0. The tumor growth kinetics of mice in the “HCA–SS–HCPT NPs without light” cohort is similar to that of the “free HCPT” group. Dramatically, after exposure of tumors from HCA–SS–HCPT NP-treated mice to white light, excellent anticancer efficacy is achieved with only a 1.75 times increase in tumor volume on day 16 (Fig. 6B). Such an excellent anticancer effect should be ascribed to the self-synergistic oxidation therapy, that is, the ROS produced by cleaved HCA provide an oxidative environment, which makes the tumor cells sensitive to the released HCPT and enables the drugs to play a much bigger role. This in vivo therapeutic result also confirms that the “0 + 1 > 1” effect observed in in vitro cellular experiment can be realized in live animals.
Furthermore, the body weight of mice in each treatment group was also monitored during the 16 day study duration. The result as shown in Fig. 6C indicates that free HCPT administration slightly reduces the mouse body weight, whereas for other treatments, there are no noticeable body weight losses observed for the mice, implying that these treatments may not have an in vivo toxic side effect. The in vivo toxicities of different treatments were also evaluated by hematoxylin and eosin (H&E) staining of slices of liver and spleen organs, which were excised on day 16. No significant damage of the liver and spleen from mice in each group can be seen (Fig. S12, ESI†), suggesting the low toxicity and good biocompatibility of our “HCA–SS–HCPT NPs with light” strategy.
Finally, on day 16, the tumor tissues were resected after the mice were sacrificed, and were then sectioned for various staining experiments in terms of H&E, TdT-mediated dUTP-biotin nick end labeling (TUNEL) and proliferating cell nuclear antigen (PCNA). As shown in Fig. 7, the histological and immunohistochemical staining images demonstrate that the treatment of “HCA–SS–HCPT NPs with light” leads to the largest necrotic regions, the most apoptotic cells, and the least proliferated cells within tumors, confirming that it has the best in vivo antitumor efficacy from the microscopic point of view.
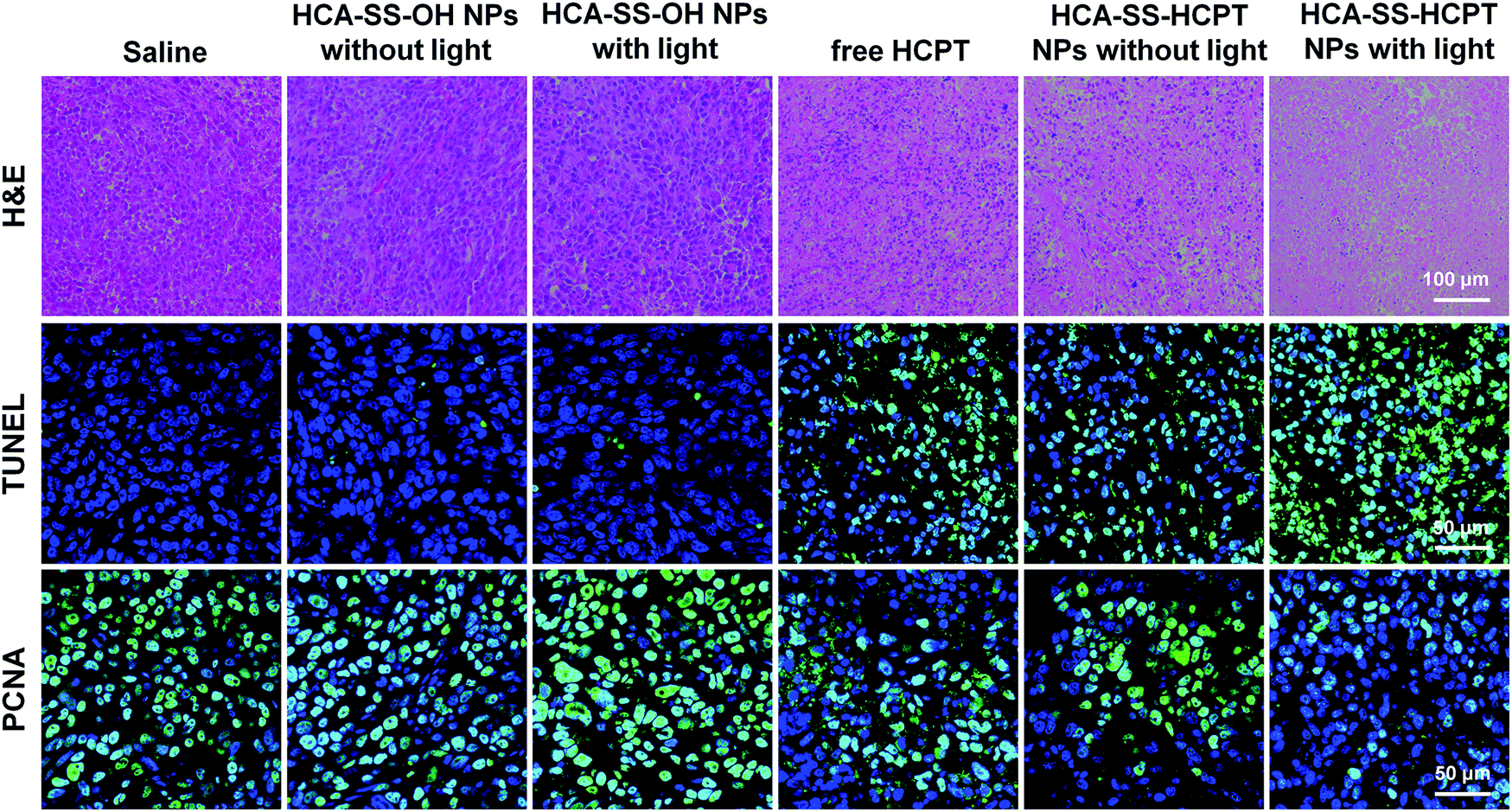 | ||
| Fig. 7 Histological and immunohistochemical observations of the tumor tissues from mice receiving different treatments on day 16. The cell nuclei were stained by DAPI for TUNEL and PCNA staining images. | ||
Conclusions
In summary, we have developed HCA–SS–HCPT NPs with a fluorescence and photoactivity dual-activatable signature. Due to the HCPT conjugation resulting in a quenching effect on HCA, HCA–SS–HCPT NPs are very weakly emissive and have negligible ROS generation ability in aqueous media. On meeting thiols such as GSH, the disulfide linker in HCA–SS–HCPT is cleaved, leading to release of intact HCPT and HCA along with intramolecular cyclization reaction. The fluorescence and ROS generation capacity of HCA hence recover, benefitting it to (1) self-report the release, location and action of the released HCPT drug; and (2) produce a small amount of ROS upon light exposure, which is not high enough to cause cancer cell death, but provides an oxidative environment inside cancer cells for self-synergistic enhancement of the anticancer effect of the released HCPT through oxidation therapy. The HCA–SS–HCPT NPs feature a co-delivery system, which allows the activated HCA and HCPT to remain in close proximity, permitting in situ reporting of the drug fate and offering an oxidative environment where the HCPT is. Moreover, the HCA–SS–HCPT NPs are thiol-responsive within cancer cells, which undoubtedly reduces the toxic side effects against normal organs as compared to free HCPT. The cellular and in vivo studies indicate that after responsiveness of HCA–SS–HCPT NPs to the high GSH level in cancer cells, the ROS produced by the cleaved HCA with white light irradiation can help the released HCPT kill many more tumor cells via yielding an intracellular oxidative environment, which achieves the self-synergistic enhancement of anticancer activity with the effect of “0 + 1 > 1”. As compared to the currently reported “dye–SS–drug” systems that mostly focused on using the dye to track the thiol-responsive drug release, this work provides a new idea of using the recovered ROS generation ability of the dye to address the resistance/insensitivity issue of cancer cells toward drugs, which will inspire the development of more exciting materials, strategies and insights in the research fields of biomedicine and cancer theranostics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (51622305, 51873092, 81773281 and 81670624), the National Basic Research Program of China (2015CB856503), and the open fund of State Key Laboratory of Luminescent Materials and Devices in South China University of Technology (2018-skllmd-05).Notes and references
- W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566 CrossRef CAS PubMed; G. Szakács, M. D. Hall, M. M. Gottesman, A. Boumendjel, R. Kachadourian, B. J. Day, H. Baubichon-Cortay and A. Di Pietro, Chem. Rev., 2014, 114, 5753 CrossRef PubMed.
- H. Wang, Z. Gao, X. Liu, P. Agarwal, S. Zhao, D. W. Conroy, G. Ji, J. Yu, C. P. Jaroniec, Z. Liu, X. Lu, X. Li and X. He, Nat. Commun., 2018, 9, 562 CrossRef PubMed.
- H. Wang, Z. Feng, Y. Qin, J. Wang and B. Xu, Angew. Chem., Int. Ed., 2018, 57, 4931 CrossRef CAS PubMed.
- S. Cross-Knorr, S. Lu, K. Perez, S. Guevara, K. Brilliant, C. Pisano, P. J. Quesenberry, M. B. Resnick and D. Chatterjee, BMC Cancer, 2013, 13, 463 CrossRef PubMed.
- J. J. G. Marin, F. S. de Medina, B. Castano, L. Bujanda, M. R. Romero, O. Martinez-Augustin, R. D. Moral-Avila and O. Briz, Drug Metab. Rev., 2012, 44, 148 CrossRef CAS PubMed.
- H. Sato, K. Maeda, K. Kotake, K. Sugihara and H. Takahashi, J. Gastroenterol., 2016, 51, 465 CrossRef CAS PubMed.
- H. C. Huang, J. Liu, Y. Baglo, I. Rizvi, S. Anbil, M. Pigula and T. Hasan, Mol. Cancer Ther., 2017, 17, 508 CrossRef PubMed.
- X. Li, X. Lu, H. Xu, Z. Zhu, H. Yin, X. Qian, R. Li, X. Jiang and B. Liu, Mol. Pharmaceutics, 2012, 9, 222 CrossRef CAS PubMed.
- S. Park, B. Kwon, W. Yang, E. Han, W. Yoo, B. M. Kwon and D. Lee, J. Controlled Release, 2014, 196, 19 CrossRef CAS PubMed.
- B. Ramanathan, K. Y. Jan, C. H. Chen, T. C. Hour, H. J. Yu and Y. S. Pu, Cancer Res., 2005, 65, 8455 CrossRef CAS PubMed.
- J. Fang, T. Sawa, T. Akaike and H. Maeda, Cancer Res., 2002, 62, 3138 CAS.
- W. Yin, J. Li, W. Ke, Z. Zha and Z. Ge, ACS Appl. Mater. Interfaces, 2017, 9, 29538 CrossRef CAS PubMed.
- H. Qiao, D. Fang, L. Zhang, X. Gu, Y. Lu, M. Sun, C. Sun, Q. Ping, J. Li, Z. Chen, J. Chen, L. Hu and L. Di, ACS Appl. Mater. Interfaces, 2018, 10, 4569 CrossRef CAS PubMed.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488 RSC.
- A. Kamkaew, F. Chen, Y. Zhan, R. L. Majewski and W. Cai, ACS Nano, 2016, 10, 3918 CrossRef CAS PubMed.
- J. Qi, C. Chen, X. Zhang, X. Hu, S. Ji, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, Nat. Commun., 2018, 9, 1848 CrossRef PubMed.
- M. Li, Z. Luo and Y. Zhao, Sci. China: Chem., 2018, 61, 1214 CrossRef CAS.
- Y. Yuan, C. J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780 CrossRef CAS PubMed.
- C. Chen, Z. Song, X. Zheng, Z. He, B. Liu, X. Huang, D. Kong, D. Ding and B. Z. Tang, Chem. Sci., 2017, 8, 2191 RSC.
- H. Gao, X. Zhang, C. Chen, K. Li and D. Ding, Adv. Biosyst., 2018, 2, 1800074 CrossRef.
- J. Qi, C. Chen, D. Ding and B. Z. Tang, Adv. Healthcare Mater., 2018, 7, 1800477 CrossRef PubMed.
- X. Gu, X. Zhang, H. Ma, S. Jia, P. Zhang, Y. Zhao, Q. Liu, J. Wang, X. Zheng, J. W. Y. Lam, D. Ding and B. Z. Tang, Adv. Mater., 2018, 30, 1801065 CrossRef PubMed.
- X. Ni, X. Zhang, X. Duan, H. L. Zheng, X. S. Xue and D. Ding, Nano Lett., 2019, 19, 318 CrossRef CAS PubMed.
- J. Qi, Y. Fang, R. T. K. Kwok, X. Zhang, X. Hu, J. W. Y. Lam, D. Ding and B. Z. Tang, ACS Nano, 2017, 11, 7177 CrossRef CAS PubMed.
- C. Y. Y. Yu, H. Xu, S. Ji, R. T. K. Kwok, J. W. Y. Lam, X. Li, S. Krishnan, D. Ding and B. Z. Tang, Adv. Mater., 2017, 29, 1606167 CrossRef PubMed.
- Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- K. Li, D. Ding, Q. Zhao, J. Sun, B. Z. Tang and B. Liu, Sci. China: Chem., 2013, 56, 1228 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- J. Li, Z. Zhu, S. Rong, H. Li, Y. Guo, Q. Xue and D. Ding, Biomater. Sci., 2017, 5, 1622 RSC.
- Z. Song, R. T. K. Kwok, E. Zhao, Z. He, Y. Hong, J. W. Y. Lam, B. Liu and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17245 CrossRef CAS PubMed.
- W. S. Shin, S. K. Park, P. Verwilst, S. Koo, J. H. Lee, S. G. Chi and J. S. Kim, Chem. Commun., 2017, 53, 1281 RSC.
- F. Hu, Y. Yuan, D. Mao, W. Wu and B. Liu, Biomaterials, 2017, 144, 53 CrossRef CAS PubMed.
- H. Han, Q. Jin, Y. Wang, Y. Chen and J. Ji, Chem. Commun., 2015, 51, 17435 RSC.
- Y. Liu, S. Zhu, K. Gu, Z. Guo, X. Huang, M. Wang, H. M. Amin, W. Zhu and P. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 29496 CrossRef CAS PubMed.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071 CrossRef CAS PubMed.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670 RSC.
- L. Rong, C. Zhang, Q. Lei, H. L. Sun, S. Y. Qin, J. Feng and X. Z. Zhang, Chem. Commun., 2015, 51, 388 RSC.
- Q. Zhang, J. He, M. Zhang and P. Ni, J. Mater. Chem. B, 2015, 3, 4922 RSC.
- Y. Zhang, Q. Yin, J. Yen, J. Li, H. Ying, H. Wang, Y. Hua, E. J. Chaney, S. A. Boppart and J. Cheng, Chem. Commun., 2015, 51, 6948 RSC.
- X. Wu, X. Sun, Z. Guo, J. Tang, Y. Shen, T. D. James, H. Tian and W. Zhu, J. Am. Chem. Soc., 2014, 136, 3579 CrossRef CAS PubMed.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668 CrossRef CAS PubMed.
- S. Bhuniya, S. Maiti, E. J. Kim, H. Lee, J. L. Sessler, K. S. Hong and J. S. Kim, Angew. Chem., Int. Ed., 2014, 53, 4469 CrossRef CAS PubMed.
- J. Wu, R. Huang, C. Wang, W. Liu, J. Wang, X. Weng, T. Tian and X. Zhou, Org. Biomol. Chem., 2013, 11, 580 RSC.
- F. Zhang, G. Zhu, O. Jacobson, Y. Liu, K. Chen, G. Yu, Q. Ni, J. Fan, Z. Yang, F. Xu, X. Fu, Z. Wang, Y. Ma, G. Niu, X. Zhao and X. Chen, ACS Nano, 2017, 11, 8838 CrossRef CAS PubMed.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066 CrossRef CAS PubMed.
- F. Kong, Z. Liang, D. Luan, X. Liu, K. Xu and B. Tang, Anal. Chem., 2016, 88, 6450 CrossRef CAS PubMed.
- S. Maiti, N. Park, J. H. Han, H. M. Jeon, J. H. Lee, S. Bhuniya, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 4567 CrossRef CAS PubMed.
- C. Yan, Z. Guo, Y. Shen, Y. Chen, H. Tian and W. H. Zhu, Chem. Sci., 2018, 9, 4959 RSC.
- M. Ye, X. Wang, J. Tang, Z. Guo, Y. Shen, H. Tian and W. H. Zhu, Chem. Sci., 2016, 7, 4958 RSC.
- C. Yan, Z. Guo, Y. Liu, P. Shi, H. Tian and W. H. Zhu, Chem. Sci., 2018, 9, 6176 RSC.
- S. D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496 CrossRef CAS PubMed.
- Z. Song, D. Mao, S. H. P. Sung, R. T. K. Kwok, J. W. Y. Lam, D. Kong, D. Ding and B. Z. Tang, Adv. Mater., 2016, 28, 7249 CrossRef CAS PubMed.
- J. V. Jokerst and S. S. Gambhir, Acc. Chem. Res., 2011, 44, 1050 CrossRef CAS PubMed.
- J. E. Lemaster and J. V. Jokerst, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1404 CrossRef.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CAS.
- A. Varela-Moreira, Y. Shi, M. H. A. M. Fens, T. Lammers, W. E. Hennink and R. M. Schiffelers, Mater. Chem. Front., 2017, 1, 1485 RSC.
- X. Xu, X. Ma, Y. E. Gao, M. Hou, P. Xue, C. M. Li and Y. Kang, Mater. Chem. Front., 2017, 1, 1257 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00081j |
| ‡ J. L. and X. N. contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |