
Supramolecular nanocatalyst in water: successive click-driven assembly of click-derived rod amphiphiles†
Inhye
Kim
 ab and
Eunji
Lee
*a
ab and
Eunji
Lee
*a
aSchool of Materials Science and Engineering, Gwangju Institute of Science and Technology, 123 Cheomdangwagi-ro, Buk-gu, Gwangju 61005, Republic of Korea. E-mail: eunjilee@gist.ac.kr
bGraduate School of Analytical Science and Technology, Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon 34134, Republic of Korea
First published on 26th March 2019
Abstract
A supramolecular vesicular nanocatalyst (VC) for successive CuI-catalyzed azide–alkyne cycloaddition in water was developed by self-assembly of a click chemistry-derived rod amphiphile (CA). The CA generated by click reaction of azide-terminated benzyl ether dendrons and diethynyl naphthalene self-assembles into vesicles in water. The excellent catalytic activity of the VC was endowed by CuI-chelation to triazole-containing rod block within VC wall. The VC performed click reaction of benzyl azide with phenylacetylene or trimethylsilylacetylene. The resultant hydrophobic products self-encapsulated in VC wall affect the interfacial curvature of the VC according to their structural compatibility with CA, inducing the increase in vesicular size or structural change to micelle. Interestingly, the morphology of VC-derived nanocatalyst was further controlled from micelle to vesicle and vice versa by successive click reactions, leading to controllable loading/release of hydrophilic payloads. This recyclable catalytic activity of the self-transformable nanocatalyst was confirmed by a visually detectable click reaction offering a fluorescent color change of the aqueous VC solution.
Introduction
Supramolecular nanovesicles based on the aqueous self-assembly of amphiphiles possess two types of confined spaces: the aqueous core cavity and the hydrophobic wall.1–3 The confined wall enables the vesicle to serve as a nanoreactor by providing a hydrophobic environment, where chemical reactions such as a Diels–Alder reaction,4 palladium-catalyzed carbon–carbon bond forming reaction,5,6 and enzymatic cascade reaction7–9 can be performed. This ability can be used to increase the conversion efficiency by locally concentrating the reacting species and pre-organizing them in a favored conformation for a desired reaction.10 The vesicles can also show a nanostructural change in response to external stimuli such as pH, light, temperature, and guest molecules,11–13 resulting from the interfacial curvature change of vesicles by the dynamic assembly behavior of stimuli-responsive block-containing amphiphiles.14,15 This makes them attractive candidates for developing transformable nanomaterials in smart chemo/biosensors and controlled drug/imaging agent delivery.14–17 However, self-transformable nanoreactors that can act as an on-demand nanotransporter of payloads has rarely been reported.CuI-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry, resulting in 1,4-disubstituted 1,2,3-triazole derivatives,18–20 has led to a wealth of applications due to its synthetic simplicity, mild reaction conditions, and biocompatibility.21–24 However, the instability of catalytically active CuI sites requires a large amount of CuI catalyst to accelerate the CuAAC click reaction. The complete removal of CuI is also a challenge and limits the utilization of click chemistry in electronics, biomedicine, and green chemistry.25 Interestingly, the click-derived triazole group has also been attractive as a ligand providing several donor sites for chelating metal ions,26,27 increasing its use in click chemistry with applications such as chemosensing,28 catalysis,29 and therapeutics.30,31 Therefore, one can envision that the click reaction-derived rod amphiphiles spontaneously form vesicles in water, providing a hydrophobic space where catalytic CuI is chelated by a triazole group and can act as a vesicular nanocatalyst (VC) for click reactions.
Herein, we report the supramolecular self-transformable VC based on the self-assembly of a click-derived, CuI-chelating rod amphiphile (Fig. 1). The VC can carry out successive click reactions with an exceptional conversion efficiency owing to (i) stabilized catalytic CuI and reactants preserving the catalytic activity, and (ii) the reactive species in close proximity to the catalytic sites in the VC wall. Also, the self-encapsulation of the hydrophobic click products within the wall affects the interfacial curvature and thus the morphology of the VC, enabling the extensive use of VCs as smart nanomaterials.
 | ||
| Fig. 1 Interfacial curvature-controllable supramolecular vesicular nanocatalyst (VC) for successive click I-to-click II reactions, formed by self-assembly of a click-derived rod amphiphile (CA) in water, depending on the molecular structure similarity of click reaction products P1 and P2 with the rod segment of CA. P1 and P2 are the click reaction products of benzyl azide with phenylacetylene or trimethylsilylacetylene, respectively. | ||
Results and discussion
The click-derived rod amphiphile (CA) was synthesized by the CuAAC reaction of azide-terminated benzyl ether dendrons and diethynyl naphthalene (Fig. S1–S3, ESI†) and showed vesicular formation by aqueous self-assembly (Fig. 1 and Fig. S6b, ESI†). The CuII-chelation to the triazolyl moiety of CA, to provide the CuI-chelated VC (CuI-VC), was examined by ultraviolet-visible (UV-Vis), fluorescence, and 1H nuclear magnetic resonance (NMR) spectroscopies (Fig. S6 and S7, ESI†). CuSO4 (0.5 equiv. mol to CA) and sodium ascorbate (NaAsc, 3.0 equiv. mol to CuSO4) were added to an aqueous solution of CA for the in situ reduction of CuII to CuI (Fig. 2). The stoichiometric ratio of CA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CuII was 2:1 (Fig. S6c, ESI†).32 In the UV-Vis spectra, the characteristic d–d transition of CuII in the range 700–900 nm33 disappeared, and new absorption peaks appeared at 600 and 750 nm after addition of CuSO4 to the aqueous solution of CA, which could be assigned to a metal-to-ligand-transfer transition (Fig. S6d, ESI†).34 The two peaks at 600 and 750 nm disappeared, and a band at 670 nm appeared after further reduction of CuII to CuI by NaAsc, indicating CuI-chelation by the triazolyl group of CA. The decrease in emission of CA after addition of CuSO4, indicative of the CuII-chelation by the triazole group of CA, could be attributed to the reverse photoinduced electron transfer35 involving electron donation from the excited naphthalene unit to the CuII-binding triazole group (Fig. S6e, ESI†).36 The 1H NMR signal of the triazole proton (Hc) of CA at 8.16 ppm measured in D2O shifted upfield to 8.12 ppm upon addition of CuSO4 (Fig. S7a, ESI†).25 The Hg of CA shifted upfield by 0.06 ppm, suggesting the oxygen atoms next to the phenyl group were interacting with CuII. After further addition of NaAsc, the Hc- and Hg-signals showed a downfield shift of 0.04 and 0.03 ppm, respectively, indicating CA–CuI complex formation. Fig. S7b (ESI†) shows the plausible binding model of CA–CuI. The successful fabrication of vesicular structures of CA–CuI for CuI-VC was confirmed by transmission electron microscopy (TEM) (Fig. S8, ESI†). The unstained TEM images of both vesicular CA–CuII precatalyst and CA–CuI catalyst clearly showed the vesicular wall with enhanced contrast due to the chelation of Cu by the triazole group at the hydrophobic segment.
:CuII was 2:1 (Fig. S6c, ESI†).32 In the UV-Vis spectra, the characteristic d–d transition of CuII in the range 700–900 nm33 disappeared, and new absorption peaks appeared at 600 and 750 nm after addition of CuSO4 to the aqueous solution of CA, which could be assigned to a metal-to-ligand-transfer transition (Fig. S6d, ESI†).34 The two peaks at 600 and 750 nm disappeared, and a band at 670 nm appeared after further reduction of CuII to CuI by NaAsc, indicating CuI-chelation by the triazolyl group of CA. The decrease in emission of CA after addition of CuSO4, indicative of the CuII-chelation by the triazole group of CA, could be attributed to the reverse photoinduced electron transfer35 involving electron donation from the excited naphthalene unit to the CuII-binding triazole group (Fig. S6e, ESI†).36 The 1H NMR signal of the triazole proton (Hc) of CA at 8.16 ppm measured in D2O shifted upfield to 8.12 ppm upon addition of CuSO4 (Fig. S7a, ESI†).25 The Hg of CA shifted upfield by 0.06 ppm, suggesting the oxygen atoms next to the phenyl group were interacting with CuII. After further addition of NaAsc, the Hc- and Hg-signals showed a downfield shift of 0.04 and 0.03 ppm, respectively, indicating CA–CuI complex formation. Fig. S7b (ESI†) shows the plausible binding model of CA–CuI. The successful fabrication of vesicular structures of CA–CuI for CuI-VC was confirmed by transmission electron microscopy (TEM) (Fig. S8, ESI†). The unstained TEM images of both vesicular CA–CuII precatalyst and CA–CuI catalyst clearly showed the vesicular wall with enhanced contrast due to the chelation of Cu by the triazole group at the hydrophobic segment.
 | ||
| Fig. 2 Development of the CuI-VC for CuAAC. | ||
The catalytic ability of CuI-VC was assessed by a 1H NMR study. First, the CuAAC reaction using water-insoluble benzyl azide (5 mol equiv. to CA for CuI-VC) and phenylacetylene (5 equiv.) was performed for 24 h to generate click product P1 in the presence of CuI-VC (Fig. S5a, ESI† and Table 1). After dissolving the resultant nanocatalyst in CDCl3, a conversion efficiency of 99% was reached (Table 1, entry 1). The reaction did not occur in the absence of CuI-VC (entry 2). The enhanced reactivity of CuI-VC was confirmed by comparison to the reaction with CuI, showing moderate catalytic conversion of 31% (entry 3). This could be attributed to a closer contact of preorganized hydrophobic reactants to stabilized catalytically active CuI sites confined in the walls of the supramolecular CuI-VC.37
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Alkyne | Time (h) | Conversionb (%) | Product name |
| a All reactions were carried out with 5 equiv. mol (12.5 μmol) of benzyl azide and alkyne in 0.5 mL distilled water at room temperature. b Reaction conversion was determined by 1H NMR spectroscopy. c CuI was prepared by addition of 3.0 equiv. NaAsc into the 0.1 mM CuSO4 solution under Ar atmosphere. | |||||
| 1 | CuI-VC |
|
24 | 99 | P1 |
| 2 | · |

|
24 | 0 | · |
| 3 | CuIc |

|
24 | 31 | P1 |
| 4 | CuI-VC |

|
24 | 98 | P2 |
| 5 | · |

|
24 | 0 | · |
| 6 | P2-loaded CuI-VC |
|
24 | 99 | P1 |
| 7 | P1-loaded CuI-VC |

|
24 | 36 | P2 |
Consequently, the water-insoluble click product P1 was generated and self-encapsulated in the hydrophobic vesicular membrane. The 1H NMR spectrum of P1-loaded CuI-VC in D2O showed the upfield shift of naphthyl protons (Hd, He, and Hf, around 0.10–0.14 ppm), benzylic Hb (0.05 ppm), and triazolyl Hc (0.06 ppm) protons of CuI-VC compared with that of the original one in D2O (Fig. S9c, ESI†). The upfield shifts of Hb, Hc, Hd, He, and Hf could be ascribed to the shield effect of P1 by the π–π stacking with the rod amphiphile CA.38 The absorbance of CuI-VC decreased and the emission was quenched after generation of P1 by the CuAAC reaction (Fig. S10, ESI† and Fig. 3a) which could be attributed to the energy transfer of CuI-VC to P1 due to the π–π stacking between the aromatic block of CA and P1.39 TEM images revealed that the vesicular structure of CuI-VC remains intact after loading P1 by the CuAAC reaction (Fig. 3b). Even in the presence of excess reactants, precipitating P1 (white solid) by the constant reactivity of CuI-VC, the vesicular structure was maintained without rupture (Fig. S11a and b, ESI†).
 | ||
| Fig. 3 (a) Emission spectra (λex = 260 nm) of CuI-VC, P1-, and P2-loaded CuI-VC in water. Negatively stained TEM image of (b) P1- and (c) P2-loaded CuI-VC (0.02 mM) with 2 wt% uranyl acetate. Insets of (b and c) show the cryo-TEM image. Inset scale bar equals (b) 100 and (c) 50 nm, respectively. (d) Hydrodynamic diameters of CuI-VC, P1-, and P2-loaded CuI-VC. | ||
To confirm the catalytic activity of CuI-VC, P2 with a bulky trimethyl group was synthesized using the CuAAC reaction of trimethylsilylacetylene and benzyl azide (Fig. 1, Table 1 and Fig. S5b, ESI†). CuI-VC shows an excellent conversion efficiency of 98% (entry 4). When the P2-forming CuAAC reaction was carried out without CuI-VC, the reaction did not occur as expected (entry 5). The 1H NMR spectrum of CuI-VC with P2 measured in D2O showed an upfield shift similar to that observed for P1-loaded CuI-VC in D2O, indicating the encapsulation of P2 by CuI-VC (Fig. S12c, ESI†). The P2-loaded CuI-VC showed the formation of micelles of diameter ∼10 nm (Fig. 3c and Fig. S13c, ESI†), which is twice the molecular length of fully extended CA (Fig. S6a, ESI†). The emission quenching of CuI-VC upon generation of P2 suggests the intercalation of P2 within the hydrophobic wall of CuI-VC (Fig. 3a). The smaller decrease in the emission of P2-loaded CuI-VC than that of P1-loaded CuI-VC could be the result of less effective π–π stacking of P2 with the hydrophobic rod segments of the CA nanocatalyst.40 The bulky trimethyl groups of P2 may hinder the close packing of CA within the membrane of CuI-VC, resulting in morphological transformation of CuI-VC from vesicle to micelle. Dynamic light scattering (DLS) revealed an increase in the average hydrodynamic diameter (RH) of P1-intercalated CuI-VC (from ∼21 nm to ∼600 nm) (Fig. 3d). A decrease in the RH of P2-intercalated CuI-VC was observed (from ∼21 nm to ∼10 nm) compared with CuI-VC. Therefore, the molecular structure similarity of the click reaction products with the hydrophobic rod segment of CA may affect the interfacial curvature of supramolecular nanocatalyst CuI-VC, resulting in a morphological change. For instance, the high structural compatibility of P1 with CA could enable the effective encapsulation of P1 within the membrane of CuI-VC. The π–π stacking of P1 with CA could be optimized as P1 and the rigid hydrophobic segment of CA are arranged in parallel, which decreased the interfacial curvature of the wall, leading to an increase in CuI-VC size.41 These results are consistent with the entropic cost of intercalating molecules with increased volume, which is responsible for the vesicle-to-micelle morphological change.42
As the interfacial curvature of CuI-VC can be tuned by the molecular architecture of click products, we studied whether the reversible morphological transformation of nanocatalyst from micellar aggregates to vesicles could be achieved by sequential click reactions to form P1 using P2-loaded CuI-VC (Fig. 4a). The CuAAC reaction of benzyl azide and phenylacetylene catalyzed by P2-loaded CuI-VC was performed (Table 1, entry 6). The catalytic activity of the P2-loaded nanoreactor still shows a tremendous conversion efficiency of 99% despite the pre-loading of P2 in CuI-VC. In the 1H NMR spectrum of P2-loaded CuI-VC after the CuAAC reaction to form P1 in D2O, Hb, Hc, Hd, He, and Hf are significantly downfield shifted (0.05–0.09 ppm), probably indicating a strong hydrogen bonding interaction between the triazolyl and naphthyl protons of CA and the aromatic protons and nitrogen atoms in the triazole moiety of P1 and P2 (Fig. 4b).38 Ha and Hg showed a moderate downfield shift (0.05 ppm), which suggests the enhanced hydrogen bonding interaction of polyethylene oxide chains of CA with water. However, after dissolving the resultant nanocatalyst in CDCl3, only P1 signals were detected in the 1H NMR spectrum (Fig. 4b and Fig. S14, ESI†). Fluorescence quenching of P2-loaded CuI-VC was observed after the P1-forming CuAAC reaction (Fig. S15b, ESI†). The TEM experiment showed the presence of vesicles (Fig. 4c). These results can be explained by preloaded P2 in CuI-VC being replaced with P1 and released into the aqueous solution because P1 has greater molecular structure compatibility with CA, which can form a more stable vesicle through π-stacking. The released P2 to the aqueous phase was confirmed by 1H NMR experiment. The transformation of P2-loaded nanocatalyst to a vesicular structure by successive click reactions was further confirmed using hydrophilic dye fluorescein isothiocyanate (FITC) that can be encapsulated in the interior cavity of the vesicle (Fig. S16a, ESI†).43 As expected, the fluorescence micrograph clearly showed bright green-emitting spherical aggregates, indicating the formation of a vesicle (Fig. 4d and Fig. S16b, ESI†).
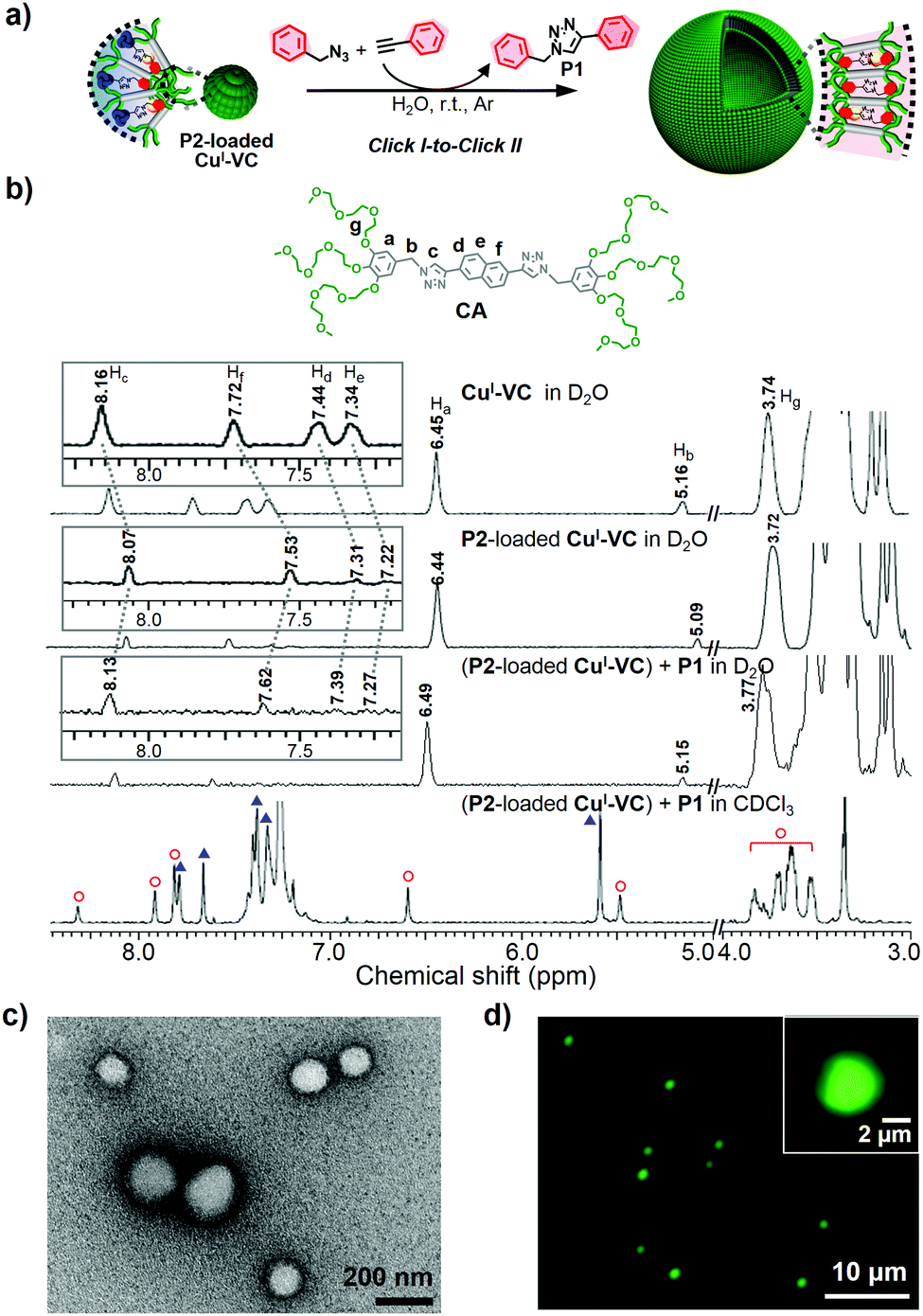 | ||
Fig. 4 (a) Successive click reactions of nanocatalyst, resulting in the morphological change of CuI-VC from micelle to vesicle. (b) 1H NMR spectra of CuI-VC and P2-loaded CuI-VC measured in D2O and P2-loaded CuI-VC after click reaction to generate P1 measured in D2O and CDCl3. Red hollow circle ( ) and blue triangle ( ) and blue triangle ( ) denote CA and P1, respectively. More detailed information is in the ESI.† (c) Negatively stained TEM image and (d) fluorescence micrograph of the P2-loaded CuI-VC after click reaction to synthesize P1. Inset of (d) shows the vesicle containing FITC within the inner aqueous medium separated by a wall from the outer aqueous solution. Green fluorescence: FITC. ) denote CA and P1, respectively. More detailed information is in the ESI.† (c) Negatively stained TEM image and (d) fluorescence micrograph of the P2-loaded CuI-VC after click reaction to synthesize P1. Inset of (d) shows the vesicle containing FITC within the inner aqueous medium separated by a wall from the outer aqueous solution. Green fluorescence: FITC. | ||
The morphological transformation of P1-loaded nanocatalyst was also demonstrated by conducting P2-forming CuAAC reaction (Fig. 5, Fig. S14 and S17, ESI†). The P1-loaded vesicular nanocatalyst encapsulating FITC in inner aqueous compartment could release the FITC fluorescent dyes into the outer aqueous phase after performing a P2-forming CuAAC reaction as a result of the nanostructural transformation to micelle (Fig. 5b). The TEM image clearly showed the micellar structures with a diameter of ∼10 nm (Fig. 5c). The 1H NMR spectrum of the resultant micelles dissolved in CDCl3 indicated the presence of both P1 and P2 within the micelle core (Fig. S14, ESI†) which could be because the preloaded P1 is not fully replaced with P2 due to the higher structural compatibility of P1 with CA. The conversion efficiency of P2 production using P1-loaded CuI-VC was calculated to be relatively low (36%) (Table 1, entry 7). This result could be attributed to the strong mutual interaction between CA and P1, limiting the access of trimethylsilylacetylene and benzyl azide to the catalytic CuI located in the vesicular wall. Therefore, the molecular packing constraint induced by preloaded products within the hydrophobic wall of the nanocatalyst should be considered for performing successive click reactions.
 | ||
| Fig. 5 (a) Morphological transformation of CuI-VC from vesicle to micelle by successive click reactions of nanocatalyst, leading to controllable loading/release of FITC payloads. (b) Emission enhancement of the FITC-released aqueous solution of P1-loaded CuI-VC (0.5 mM) incorporating FITC after click reaction to form P2. (c) Negatively stained TEM image of P1-loaded CuI-VC after P2-forming CuAAC reaction with 2 wt% uranyl acetate. | ||
The recyclable catalytic activity of CuI-VC with structural transformable capability was further confirmed by conducting a visually detectable click reaction via a fluorescent color change of a CuI-VC solution. The conversion of water-insoluble nonfluorescent reactants to fluorescent products by the CuAAC reaction can help detect the click reaction using the naked eye. The hydrophobic coumarin derivatives were utilized as a target click product. Upon adding coumarin into aqueous CuI-VC solution, characteristic absorption of coumarin (∼280 nm) appeared while the fluorescence of CuI-VC was quenched, indicating the presence of coumarin within the hydrophobic segment of CuI-VC (Fig. S18, ESI†). To generate a coumarin-based fluorescent click product with different structural similarities to CA, we synthesized 3-azido-7-hydroxycoumarin, a nonfluorescent azide (Fig. S4, ESI†).44,45 The fluorescent emission of the coumarin moiety in 3-azido-7-hydroxycoumarin is quenched by the lone pair of electrons from the azido group.46,47 Phenylacetylene and trimethylsilylacetylene were also used for the CuAAC reactions with 3-azido-7-hydroxycoumarin (Fig. 6a). The resulting click products of phenylacetylene and trimethylsilylacetylene with 3-azido-7-hydroxycoumarin were referred to as CP1 and CP2, respectively (Fig. 6a). As expected, the blue color of 5 mM CuI-VC solution became turquoise under irradiation at 365 nm, indicating the formation of CP1 and the subsequent loading within the CuI-VC vesicular membrane. In contrast, the CP2-containing CuI-VC solution showed relatively weak green emission under UV irradiation. A new absorption band around 340 nm appeared for both CP1- and CP2-loaded CuI-VC (Fig. 6b), and the fluorescence of CuI-VC was quenched (Fig. 6c). These results indicate that click products CP1 and CP2 were incorporated within the CuI-VC wall. Notably, a characteristic emission peak of CP1 at 480 nm appeared for CP1-loaded CuI-VC when excited at the wavelength of 260 nm which indicates the fluorescence resonance energy transfer (FRET) due to the close proximity of CP1 and the rod block of CA.48 The electron lone pair of the azido moiety of 3-azido-7-hydroxycoumarin could be localized as the triazole ring is formed in CP1, leading to the activation of fluorescence.46,47 The emergence of the FRET signal demonstrated efficient stacking between CP1 and the rod amphiphile CA. The fluorescence micrograph of the CP1-loaded CuI-VC confirmed the presence of spherical structures (Fig. 6d), and TEM revealed the intact vesicular structure of CP1-encapsulated CuI-VC (inset of Fig. 6d and Fig. S19, ESI†). Loading of CP2 with an unsymmetric structure imposed by the bulky trimethyl group into CuI-VC showed the formation of the micellar aggregates (Fig. 6e). These results are consistent with the morphological transition from vesicles to micelles observed for CuI-VC when synthesizing P1 and P2 based on the benzyl azide.
 | ||
| Fig. 6 (a) CuI-VC nanocatalyst with images of a 5 mM CuI-VC solution under UV irradiation (λex = 365 nm). Change in (b) absorption and (c) emission spectra of 0.02 mM CuI-VC upon CuAAC reaction to generate CP1 and CP2, respectively (λex = 260 nm). (d) Fluorescence micrograph of CP1-loaded CuI-VC. Inset shows the negatively stained TEM image of CP1-loaded CuI-VC with a scale bar of 50 nm. (e) Negatively stained TEM image of CP2-incorporated CuI-VC. Inset shows the diameter distribution of the micelle. | ||
Conclusions
In summary, we have developed an aqueous supramolecular nanocatalyst based on the self-assembly of the click-derived rod amphiphile, capable of successive click reactions, which maintained excellent catalytic efficiency. The self-transformable behavior of the nanocatalyst was controlled by encapsulating structurally compatible hydrophobic click reaction products with a rod amphiphile, which, in turn, affects the interfacial curvature of the nanocatalyst. The morphological transformability enables the click nanoreactor/catalyst potential for in situ production of on demand payloads and controllable loading/release of such payloads, including drugs, imaging agents, and chemical species. This can further broaden the potential applications of supramolecular nanocatalysts into the field of smart nanobiomedicine and chemo/biosensors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by Basic Science Research Program (2016R1A2B4012322) through the National Research Foundation of Korea (NRF). We thank Prof. B. K. Cho for the discussion of amphiphile CA synthesis.References
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049 CrossRef CAS PubMed.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- T. Rispens and J. B. F. N. Engberts, Org. Lett., 2001, 3, 941–943 CrossRef CAS PubMed.
- G. Hamasaka, T. Muto and Y. Uozumi, Angew. Chem., Int. Ed., 2011, 50, 4876–4878 CrossRef CAS PubMed.
- M. Liu, X. Zhu, L. Wu, X. Zhou, J. Li and J. Ma, RSC Adv., 2015, 5, 38264–38270 RSC.
- R. J. R. W. Peters, I. Louzao and J. C. M. van Hest, Chem. Sci., 2012, 3, 335–342 RSC.
- D. M. Vriezema, P. M. L. Garcia, N. S. Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS PubMed.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. R. de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS PubMed.
- D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- L. Jiang, X. Huang, D. Chen, H. Yan, X. Li and X. Du, Angew. Chem., Int. Ed., 2017, 56, 2655–2659 CrossRef CAS PubMed.
- W. Zhang and C. Gao, J. Mater. Chem. A, 2017, 5, 16059–16104 RSC.
- Z.-Q. Cao, Y.-C. Wang, A.-H. Zou, G. London, Q. Zhang, C. Gao and D.-H. Qu, Chem. Commun., 2017, 53, 8683–8686 RSC.
- M.-H. Li and P. Keller, Soft Matter, 2009, 5, 927–937 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- J.-F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS PubMed.
- D. S. Tyler, J. Vappiani, T. Cañeque, E. Y. N. Lam, A. Ward, O. Gilan, Y.-C. Chan, A. Hienzsch, A. Rutkowska, T. Werner, A. J. Wagner, D. Lugo, R. Gregory, C. R. Molina, N. Garton, C. R. Wellaway, S. Jackson, L. MacPherson, M. Figueiredo, S. Stolzenburg, C. C. Bell, C. House, S.-J. Dawson, E. D. Hawkins, G. Drewes, R. K. Prinjha, R. Rodriguez, P. Grandi and M. A. Dawson, Science, 2017, 356, 1397–1401 CrossRef CAS PubMed.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS PubMed.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed.
- H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675–696 RSC.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- A. Maisonial, P. Serafin, M. Traïkia, E. Debiton, V. Théry, D. J. Aitken, P. Lemoine, B. Viossat and A. Gautier, Eur. J. Inorg. Chem., 2008, 298–305 CrossRef CAS.
- M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. W. Yong, S. Tsutsui, P. K. Stys and T. Storr, Chem. Sci., 2017, 8, 5636–5643 RSC.
- I. Kim, N.-E. Lee, Y.-J. Jeong, Y.-H. Chung, B.-K. Cho and E. Lee, Chem. Commun., 2014, 50, 14006–14009 RSC.
- H. Irie, K. Kamiya, T. Shibanuma, S. Miura, D. A. Tryk, T. Yokoyama and K. Hashimoto, J. Phys. Chem. C, 2009, 113, 10761–10766 CrossRef CAS.
- T. J. Meyer, Pure Appl. Chem., 1986, 58, 1193–1206 CAS.
- A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385–2393 RSC.
- Y.-C. Hsieh, J.-L. Chir, H.-H. Wu, P.-S. Chang and A.-T. Wu, Carbohydr. Res., 2009, 344, 2236–2239 CrossRef CAS PubMed.
- L. Qin, L. Zhang, Q. Jin, J. Zhang, B. Han and M. Liu, Angew. Chem., Int. Ed., 2013, 52, 7761–7765 CrossRef CAS PubMed.
- Y. Zhao, Y. Li, Y. Li, C. Huang, H. Liu, S.-W. Lai, C.-M. Che and D. Zhu, Org. Biomol. Chem., 2010, 8, 3923–3927 RSC.
- V. K. Praveen, C. Ranjith, E. Bandini, A. Ajayaghosh and N. Armaroli, Chem. Soc. Rev., 2014, 43, 4222–4242 RSC.
- N. K. Allampally, A. Florian, M. J. Mayoral, C. Rest, V. Stepanenko and G. Fernández, Chem. – Eur. J., 2014, 20, 10669–10678 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- R. J. Hickey, J. Koski, X. Meng, R. A. Riggleman, P. Zhang and S.-J. Park, ACS Nano, 2014, 8, 495–502 CrossRef CAS PubMed.
- M. R. Molla, P. Rangadurai, L. Antony, S. Swaminathan, J. J. de Pablo and S. Thayumanavan, Nat. Chem., 2018, 10, 659–666 CrossRef CAS PubMed.
- C. Wang, L. Lu, W. Ye, O. Zheng, B. Qiu, Z. Lin, L. Guo and G. Chen, Analyst, 2014, 139, 656–659 RSC.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS PubMed.
- Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 13096–13105 CrossRef CAS PubMed.
- C. Besanceney-Webler, H. Jiang, T. Zheng, L. Feng, D. S. del Amo, W. Wang, L. M. Klivansky, F. L. Marlow, Y. Liu and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 8051–8056 CrossRef CAS PubMed.
- M. M. Hanczyc, S. M. Fujikawa and J. W. Szostak, Science, 2003, 302, 618–622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00059c |
| This journal is © the Partner Organisations 2019 |