
Electrode engineering starting from live biomass: a ‘smart’ way to construct smart pregnant hybrids for sustainable charge storage devices†
Jian
Jiang‡
 *a,
Siyuan
Liu‡
a,
Yani
Liu
a,
Ting
Meng
a,
Lai
Ma
a,
Han
Zhang
a,
Maowen
Xu
a,
Jianhui
Zhu
*b and
Chang Ming
Li
*a
*a,
Siyuan
Liu‡
a,
Yani
Liu
a,
Ting
Meng
a,
Lai
Ma
a,
Han
Zhang
a,
Maowen
Xu
a,
Jianhui
Zhu
*b and
Chang Ming
Li
*a
aSchool of Materials and Energy, and Chongqing Key Lab for Advanced Materials and Clean Energies of Technologies, Southwest University, No. 2 Tiansheng Road, BeiBei District, Chongqing 400715, P. R. China. E-mail: jjiang@swu.edu.cn; ecmli@swu.edu.cn; Tel: +86-23-68254842
bSchool of Physical Science and Technology, Southwest University, No. 2 Tiansheng Road, BeiBei District, Chongqing 400715, P. R. China. E-mail: jhzhu@swu.edu.cn
First published on 5th March 2019
Abstract
Evolving the use of rich/renewable biomass into useful electrodes is never out-of-date for sustained energy storage utilization. Despite the fact that scalable electrode fabrication can be achieved by a general strategy of ‘direct calcination and then combination with extra active components’, making bio-derived electrodes with smart pregnant hybrid architectures in a controlled way still remains challenging. Here, we propose a preferable electrode manufacturing protocol, by initiating the engineering with fresh biomaterials via facile biochemical routes. As a paradigm study, fresh tiny yeasts are chosen as the raw material to build functionalized hybrid electrodes of Fe3O4@yeast-evolved carbon (Fe3O4@YE-C), which can serve as prominent anodes for high-rate charge storage devices. Systematic studies verify that the reaction time plays a key role in forming such integrated hybrid configurations. The optimal synergistic cooperation between the outer YE-C ‘reactor’ and inner Fe3O4 ‘nanoactives’ endows the electrodes with outstanding anodic behaviors, comprising remarkable specific capacity, prolonged cycle lifetime and superb rate capability in either half-cell or full-cell aqueous systems. Our present work confirms the feasibility of engineering electrodes starting from live biomass, thus offering a sustainable and superior route to develop advanced and applicable charge storage devices.
Introduction
A strategy for engineering abundant biomass or domestic waste into useful electrodes in a controllable and smart manner can never be out-of-date in the development of sustainable charge storage devices, since it helps to reduce both electrode fabrication cost and environmental pollution.1 To date, various renewable biomaterials (e.g., rice husks, bamboo leaves/stems, bacteria, coconut shells, fungi, etc.) have been evaluated as electrode resources to construct distinct battery/supercapacitor systems. Their derived carbonaceous products with designated micro-/nanostructures and tunable surface chemistries (e.g., heteroatom doping) can either act directly as working materials for ion adsorption/intercalation (e.g., Li+, Na+, K+, OH−, SO42−, etc.) (thus making additional pseudo-capacitance contributions), or function as electrically conducting matrix/scaffolds to load redox actives.2–5 Of these biological species, fresh tiny yeasts (average size: ∼2 μm) from food science hold great promise in energy storage applications.6 Fresh tiny yeasts are a common member of unicellular eukaryotic microorganisms with (i) great natural abundance, (ii) rapid/robust ‘budding’ reproduction capability, (iii) rich natural heteroatom doping or functional chemical groups, and (iv) appropriate size (∼200–500 nm after carbonization) for electrochemical reactions.Previous studies have fully confirmed the feasibility of evolving manifold biomaterials into useful battery/supercapacitor electrodes on a large scale.7–9 However, nearly all of them follow the same rigid utilization routine, that is: (i) direct pyrolysis of biological species into functionalized carbon (C) frameworks with specific intrinsic properties (e.g., large specific surface areas, interior hollow architecture, mesoporous nature, high conductivity, etc.), and later (ii) continued combination of derived C with foreign species (e.g., metal oxides/hydroxides/sulfides, S/Se, etc.) to form smart actives@C matrix products.8–12 Such a pregnant configuration has unique, superior features over other hybrid constructions, being capable of (i) providing good buffering of the volume variations for inner actives, (ii) protecting actives against adverse aggregations in repeated redox reactions, (iii) avoiding direct contact between the active and the electrolyte, and (iv) linking the actives and serving as conductive channels for electron transfer.13 Nevertheless, following this concept can result in several challenging issues. One major challenge is how to effectively load electrochemical actives ‘as much as possible’ into derived carbonaceous scaffolds. Normally, filling additional materials into C inner places must be aided by physical evaporation/fusion or multi-step chemical treatments that are costly, tedious and inefficient for scalable use.14–16 Besides, the actual loading amount is quite limited since most foreign actives are prone to becoming stuck/aggregated on C exterior surfaces (as in micro-/meso-pores) to reduce the surface energy,17 rather than preferentially diffusing into the core region. Thus the as-built electrodes easily suffer from capacity fading, short-cyclic lifespan and irreversibility issues in long-lasting battery/supercapacitor operations. Moreover, the hydrophobicity for C strengthens the difficulty in this hybrid generation, which is a big bottleneck for increasing the practical use of biomass. So, seeking other preferable/economical ways to transform rich biomaterials into desirable actives@C electrodes deserves systematic investigation.
Manufacturing minerals with live organisms through classic biosynthesis methods (e.g., biomineralization) is of great scientific/engineering interest. It promotes the realization of state-of-the-art materials in environmentally benign systems and also allows delicate molecular control of the phase, size and morphology of inorganic crystals by tuning the biosynthesis conditions.6 It is particularly noteworthy that, compared with the case of dense/compact inorganic crystals, chemical agents more readily pass through the cell wall/membrane by means of appropriate biochemical treatments and gain access to the body of the biomass (e.g., fungi, bacteria, viruses, etc.), leading to a yield of functionalized hybrids with the desired pregnant configurations. Here, we put forward an efficient and alternative protocol, wherein fresh tiny yeasts are selected as the initiating materials to fabricate hybrid electrodes of ferroferric oxide@yeast-evolved C (Fe3O4@YE-C) for sustained charge storage devices. By following the biosynthesis and calcination processes, Fe3O4 nanoparticles are formed in situ and tightly impregnated within YE-C spheres, the reliable ‘reactors’ for reversible electrochemical redox reactions. To make clear the factors influencing engineering on live yeast cells, systematic studies of the relationship between the reaction time and microstate variations have also been conducted. Thanks to effective combination/synergistic cooperation of the YE-C reactors and confined Fe3O4 nanoactives, the as-made products exhibit excellent anodic behaviors in aqueous electrolytes, and are capable of showing a maximum specific capacity/capacitance of ∼218.2 mA h g−1/∼759.3 F g−1, outstanding long-term cyclic endurance (very low capacity decay in a total of 3000 cycles) and prominent rate capability (retaining a capacity/capacitance of ∼103.0 mA h g−1/∼297 F g−1 even at 32 A g−1). Furthermore, to confirm their potential in real applications, we constructed full-cell supercapacitors that can output both high specific energy and power densities (with maximum values up to ∼72 W h kg−1 and ∼17 kW kg−1, respectively). This work not only provides a scalable/sustained way to convert biomaterials into useful anodes for charge storage use, but also affirms the feasibility of engineering electrodes starting from live bio-species in a wide range of fields of research.
Experimental section
Synthesis of Fe3O4@YE-C reactors
Typically, 0.2 g fresh yeast (Angel Yeast Co., Ltd) was dissolved into 10 mL distilled water under ultrasonication for 10 min to form the uniform bioemulsion. Then, 2.5 g FeCl3 (99.99%, Sinopharm Chemical Reagent Co., Ltd) pre-dissolved in 20 mL deionized water was added into this bioemulsion under magnetic stirring for 30 min at room temperature. Later, the resultant mixture solution was transferred into a 100 mL glass reagent bottle, which was sealed and placed in an electric oven at 90 °C for 1 h. When cooled down to room temperature naturally, the brownish precipitates were collected by vacuum filtration, rinsed carefully with deionized water, and dried at 80 °C in an electric oven. Finally, the collected powders were calcinated at 650 °C under Ar flow (heating rate: 10 °C min−1) for 2 h to generate the final samples of Fe3O4@YE-C.For comparison study, bare Fe3O4 nanoparticles and the conventional core–shell Fe3O4@C hybrids were also prepared according to the literature.18,19 For bare Fe3O4 samples, 0.5 g Fe2O3 nanopowders with a central size of ∼30 nm (purchased from Dongying Technology Co. Ltd; http://www.naamall.com/index.php) were put into the center of a quartz tube. Later, ∼2 mL ethylene glycol (loaded in an alumina boat) was placed at the upstream zone of the quartz tube. The furnace was then sealed, heated to ∼400 °C (ramped rate: ∼10 °C min−1) under Ar flow (50 sccm) and held for 0.5 h. For synthesis of Fe3O4@C hybrids, 0.5 g Fe2O3 nanoparticles and 0.1 g dopamine molecules were successively dispersed into 200 mL of Tris buffer solution and treated by ultrasonication for 1 h. The intermediates of Fe2O3@polydopamine were then separated by vacuum filtration, washed with deionized water several times, and further heated in Ar atmosphere at 650 °C for 1 h.
Synthesis of α-Co(OH)2 nanowire arrays (NWs)
A 50 mL homogeneous solution containing 0.8 g Co(NO3)2·6H2O, 0.35 g NH4F and 0.8 g Co(NH2)2 was transferred into a 100 mL Teflon autoclave. Later, a piece of graphite sheet (45 × 15 × 5 mm3) was immersed into this mixed solution and placed against the liner wall. The autoclave was then sealed and heated at 130 °C in an electric oven for 6 h. After cooling down to room temperature naturally, the graphite sheet with violet samples grown on it was taken out, cleaned by ultrasonication in distilled water for a few seconds to remove the residual nanoparticle debris and dried at 60 °C in the electric oven.Characterization and electrochemical measurements
The morphology of the samples was characterized by a JEOL JSM-7800F field emission scanning electron microscope (FE-SEM) with energy-dispersive X-ray spectroscopy (EDS) and a JEM 2010F high-resolution transmission electron microscope (HR-TEM). X-ray powder diffraction (XRD) patterns were measured on a Bruker D8 Advance diffractometer using Cu-Kα radiation. X-ray photoelectron spectroscopy (XPS; Thermo Electron, VG ESCALAB 250 spectrometer) was also used to characterize the as-made products. Thermogravimetric analysis was carried out on a SDT600 apparatus with a heating rate of 10 K min−1 in air atmosphere. Measurement of the N2 adsorption/desorption (TriStar II 3020) was also conducted to characterize the specific surface areas of the samples (calculated by Brunauer–Emmett–Teller method) and pore-size distribution.All anodes were fabricated by the conventional slurry-coating method. The active electrode powders, poly(vinylidene fluoride) (PVDF) binder, and acetylene black were mixed in a mass ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 and dispersed/homogenized in N-methyl-2-pyrrolidone (NMP) to form a slurry. The homogeneous slurry was then pasted onto a Ni foam (thickness: 1.5 mm) and dried at 100 °C for 10 h under vacuum. The electrode mass was measured on a microbalance with an accuracy of 0.01 mg (Ohaus, USA). The mass loading density for Fe3O4@YE-C actives on each current collector was controlled at the level of ∼3–4.5 mg cm−2. For the matched cathode, the as-grown α-Co(OH)2 NWs on the graphite sheet can be directly used as the working electrodes. The actives mass calculation was made according to weight changes induced by the removal of α-Co(OH)2via a hydrochloric acid treatment (loading density range: ∼2.5–4.2 mg cm−2). Electrochemical measurements were all performed using a CS310 electrochemical workstation. For the half-cell testing, the working electrodes were evaluated in a three-electrode system, with a Pt foil as the counter electrode and an Ag/AgCl as the reference electrode. The full-cell supercapacitors were built with the as-made working anodes and pre-activated (by continuous cyclic voltammetry scans toward electrodes at a rate of 30 mV s−1 for ∼50 cycles) α-Co(OH)2 NWs cathode in opposition to each other in 3 M KOH aqueous electrolyte. To balance the charge storage and ensure full utilization of actives in the anode, the cathode/anode mass ratio was eventually determined to be 1.5:1 by referring to electrode behaviors in half-cell testing. The specific stored charge (Qspec.) and gravimetric capacitance (C) can be calculated based on the galvanostatic charge/discharge curves according to the eqn (1) and (2):
:10:10 and dispersed/homogenized in N-methyl-2-pyrrolidone (NMP) to form a slurry. The homogeneous slurry was then pasted onto a Ni foam (thickness: 1.5 mm) and dried at 100 °C for 10 h under vacuum. The electrode mass was measured on a microbalance with an accuracy of 0.01 mg (Ohaus, USA). The mass loading density for Fe3O4@YE-C actives on each current collector was controlled at the level of ∼3–4.5 mg cm−2. For the matched cathode, the as-grown α-Co(OH)2 NWs on the graphite sheet can be directly used as the working electrodes. The actives mass calculation was made according to weight changes induced by the removal of α-Co(OH)2via a hydrochloric acid treatment (loading density range: ∼2.5–4.2 mg cm−2). Electrochemical measurements were all performed using a CS310 electrochemical workstation. For the half-cell testing, the working electrodes were evaluated in a three-electrode system, with a Pt foil as the counter electrode and an Ag/AgCl as the reference electrode. The full-cell supercapacitors were built with the as-made working anodes and pre-activated (by continuous cyclic voltammetry scans toward electrodes at a rate of 30 mV s−1 for ∼50 cycles) α-Co(OH)2 NWs cathode in opposition to each other in 3 M KOH aqueous electrolyte. To balance the charge storage and ensure full utilization of actives in the anode, the cathode/anode mass ratio was eventually determined to be 1.5:1 by referring to electrode behaviors in half-cell testing. The specific stored charge (Qspec.) and gravimetric capacitance (C) can be calculated based on the galvanostatic charge/discharge curves according to the eqn (1) and (2):
| Qspec. = I × t/3.6m | (1) |
| C = I × t/(ΔV × m) | (2) |
| E = CV2/7.2 | (3) |
| P = E·3600/t | (4) |
Results and discussion
Fig. 1a schematically shows the synthetic flow of Fe3O4@YE-C reactors, using fresh yeast cells as the starting materials that function as both C sources and structural templates. After sufficient immersion in concentrated FeCl3 solution (with a high Fe3+ concentration gradient) and a subsequent heating process (wherein decomposition and carbonization of the inorganics proceed), tiny yeasts can successfully evolve into Fe3O4@YE-C hybrid products. Fig. 1b shows a comparison between the traditional fabrication procedures and our approach. In the traditional procedure, the original step is straightforward carbonization of dispersed yeasts to obtain C frameworks with large specific surface areas, extraordinary intrinsic nature (e.g., electrically conducting, highly porous), as well as designated heteroatom doping properties.20 To achieve high charge storage capacity, extra substances with remarkable pseudo-capacitive/battery-type Faradaic behaviors are further loaded, forming an optimal hybrid construction.2,20–22 Nevertheless, effective incorporation of foreign actives to the interior biocarbon regions is rather difficult. The main challenge stems from the fact that the YE-C (partially graphitized/crystallized) is dense and compact after high-temperature treatment. Thereby, the majority of chemicals are merely apt to deposit/accumulate on the YE-C outer surfaces rather than fill into the core spaces. Despite the fact that the as-formed electrodes can also exhibit redox behaviors when utilized in battery/supercapacitor devices, their operation parameters (e.g., reversibility, energy densities, charge storage retention and cyclic lifespan) are often far from satisfactory since the anchored actives peel off from YE-C in deep cycling. In sharp contrast, our biosynthesis concept is preferable and controllable for the following reasons: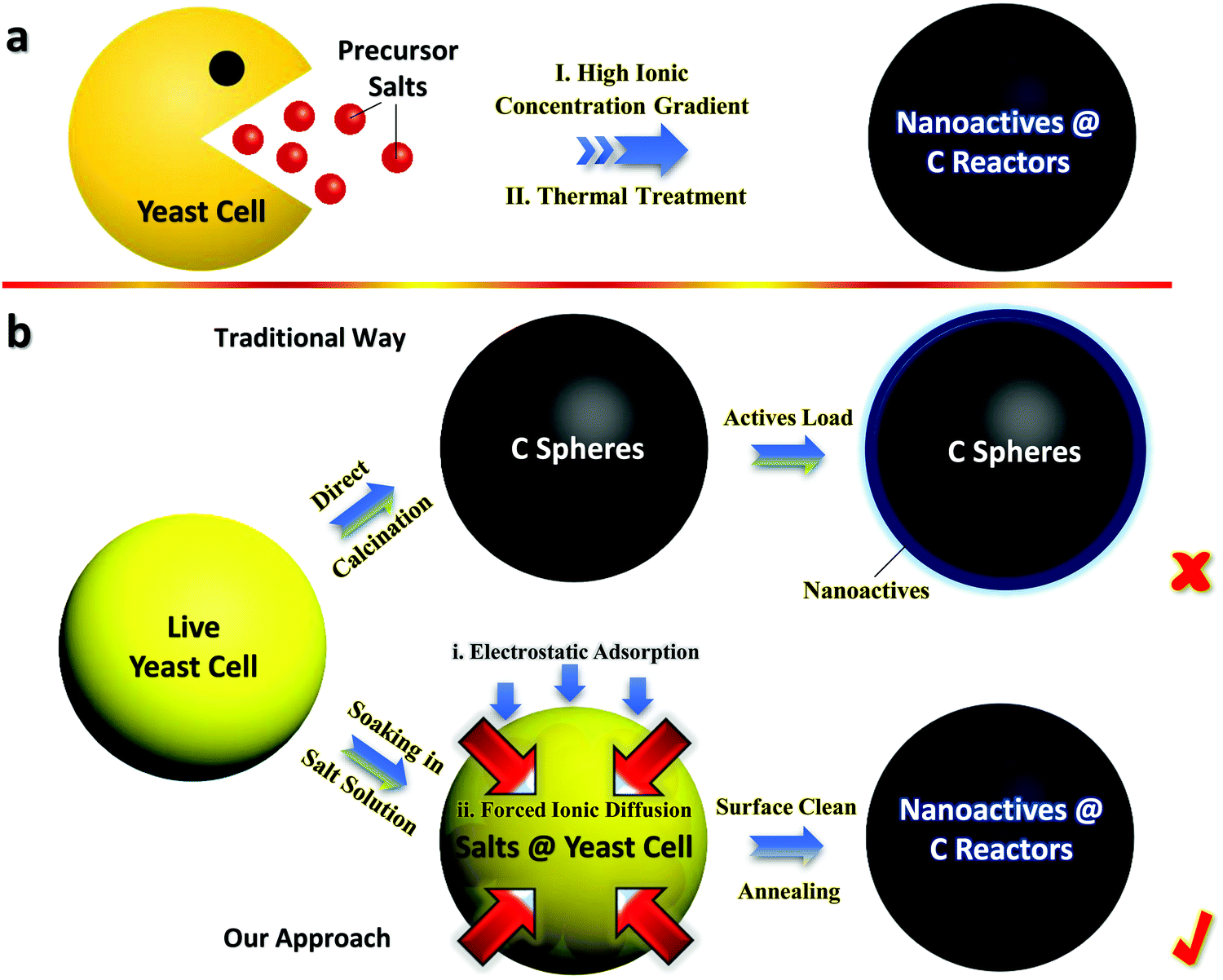 | ||
| Fig. 1 General schematics for (a) the fabrication flow of Fe3O4@YE-C reactors and (b) typical comparisons between traditional and our synthetic procedures. | ||
1. Impregnation of actives into YE-C can be readily achieved via a forced ion-diffusion method. Plenty of Fe3+ can pass through the cytoplasmic membrane and enter into yeasts, driven by the high ionic concentration gradient. Compared with tough/compact YE-C, live cells are much easier to manipulate/configure. Significantly, the overall synthesis is environmentally benign and extendable, showing great potential in sustainable charge storage fields.
2. Bioactive tiny yeasts have the ability to absorb positively charged ions, in that there are large quantities of negative groups (e.g., OH− and COO−) involved in the whole yeast body.6,23 As a result, positive ions (such as Fe3+) are prone to be gathered/enriched surrounding each cell unit, giving rise to a large increase in Fe3+ amount and the promotion/acceleration of ionic inward diffusion.
3. The configured pregnant hybrid architecture results in superior characteristics for as-built Fe3O4@YE-C electrodes over hybrid examples made by conventional methods, such as the prominent operation stability and reversibility, prolonged cyclic endurance as well as admirable rate capabilities for charge storage applications.
Typical microscopic observation of fresh tiny yeasts (Fig. 2a) uncovers their uniform spherical or elliptical morphology (diameter range: ∼1–2 μm). SEM and TEM observations (Fig. 2b and c) further confirm their unique geometric dimensions and textures (e.g., smooth exterior surface). The ultimate Fe3O4@YE-C reactors are produced after the soaking and calcination procedures. The top-view SEM image (Fig. 2d) reveals that the hybrid products still preserve their near-spherical shape but their size evidently shrinks to a level of ∼200–500 nm; the particle size decrease would be highly associated with pyrolysis and losses of bio-substances. We should highlight that this diameter range is appropriate for practical uses (especially for rechargeable power sources such as lithium-ion batteries). High-resolution SEM observations (Fig. 2e and f) disclose that the surface of Fe3O4@YE-C becomes distinctly rough; the appearance of numerous wrinkle-like nanostructures could be ascribed to chemical interactions (e.g., chemical etching) between ferruginous species and biocarbons during the high-temperature process. TEM observations (Fig. 2g) clearly reveal that Fe3O4 nanoparticles are distributed in egg-like C shells. Note the fact that the YE-C shells have been partially graphited, as reflected by HR-TEM observations (Fig. 2h). XRD has been used to analyze the final Fe3O4@YE-C samples (Fig. 2i). Except for a broad diffraction peak at a 2θ value of 22.3° arising from the YE-C, all the peak signals can be well indexed to cubic Fe3O4 (JCPDS Card No. 75-0033), corresponding to the crystalline facets of (220), (311), (400), (511) and (440). EDS and elemental mapping detection on distinct individual units (marked as A, B, C; see the corresponding SEM image in Fig. 3a) confirm the homogeneous distribution of C, O, P and Fe elements in each Fe3O4@YE-C (see EDS spectra in Fig. 3b); no other elemental impurities were detected. The strong peak signal for the P element suggests YE-C reactors contain abundant phosphorous substances that could be chiefly converted from the phospholipid in the cytomembrane/organelle. The elemental mapping (Fig. 3c–e) shows the existence of Fe, O and P elements evenly distributed in the hybrid samples. To gain insight into their inner constitution, we performed a line-scan EDS analysis on a selected sample unit (Fig. 3f and g). The records for Fe, O and P elements undoubtedly confirm the uniform elemental distribution in each Fe3O4@YE-C, together with an integrated hybrid configuration. The N2 adsorption/desorption measurement was conducted as well to characterize the specific surface area and pore-size distribution of Fe3O4@YE-C (Fig. S1, ESI†). As noted, samples made of yeast-evolved C shells and dispersive/tiny Fe3O4 cores can exhibit a total specific surface area of ∼353.7 m2 g−1, with the pore size centralized at ∼2.7 and ∼7.5 nm, respectively. Although yeasts would shrink and become solid to some extent after the annealing treatment, plenty of new mesoporous/hollow structures would simultaneously appear on biocarbons or in their inner regions, capable of offering rich and convenient transport pathways for efficient ionic diffusion. Herein, one key experimental detail should be stressed in that after the soaking procedure, the brownish precipitates collected by vacuum filtration must be rinsed carefully with deionized water so as to remove the soluble FeCl3 on the biomass outer surface. To affirm this point, we purposely examined unclean samples by SEM/TEM (see Fig. S2a–c, ESI†). The SEM images reveal that nearly all yeast cells are covered by a gel-like/thick substance so that hardly find any individual cell unit can be found; the typical TEM observation even shows that there are bulky FeCl3 crystals adhering to the yeast cells. This result in turn confirms that delicate experimental control plays a significant role in Fe3O4@YE-C evolution. Besides, to check the hybrid structural configurations, we purposely removed the Fe3O4 component involved with concentrated hydrochloric acid. The SEM images (Fig. S2e and f, ESI†) reveal that a mass of bubble-like structures are definitely distributed in the remaining products. The close SEM observations of broken YE-C units illustrate that hollow cavities exist in their inner surfaces, suggesting that the evolved products possess the egg-like shell constructions, rather than that the Fe3O4 nanoparticles accumulate/adhere on YE-C exterior surface.
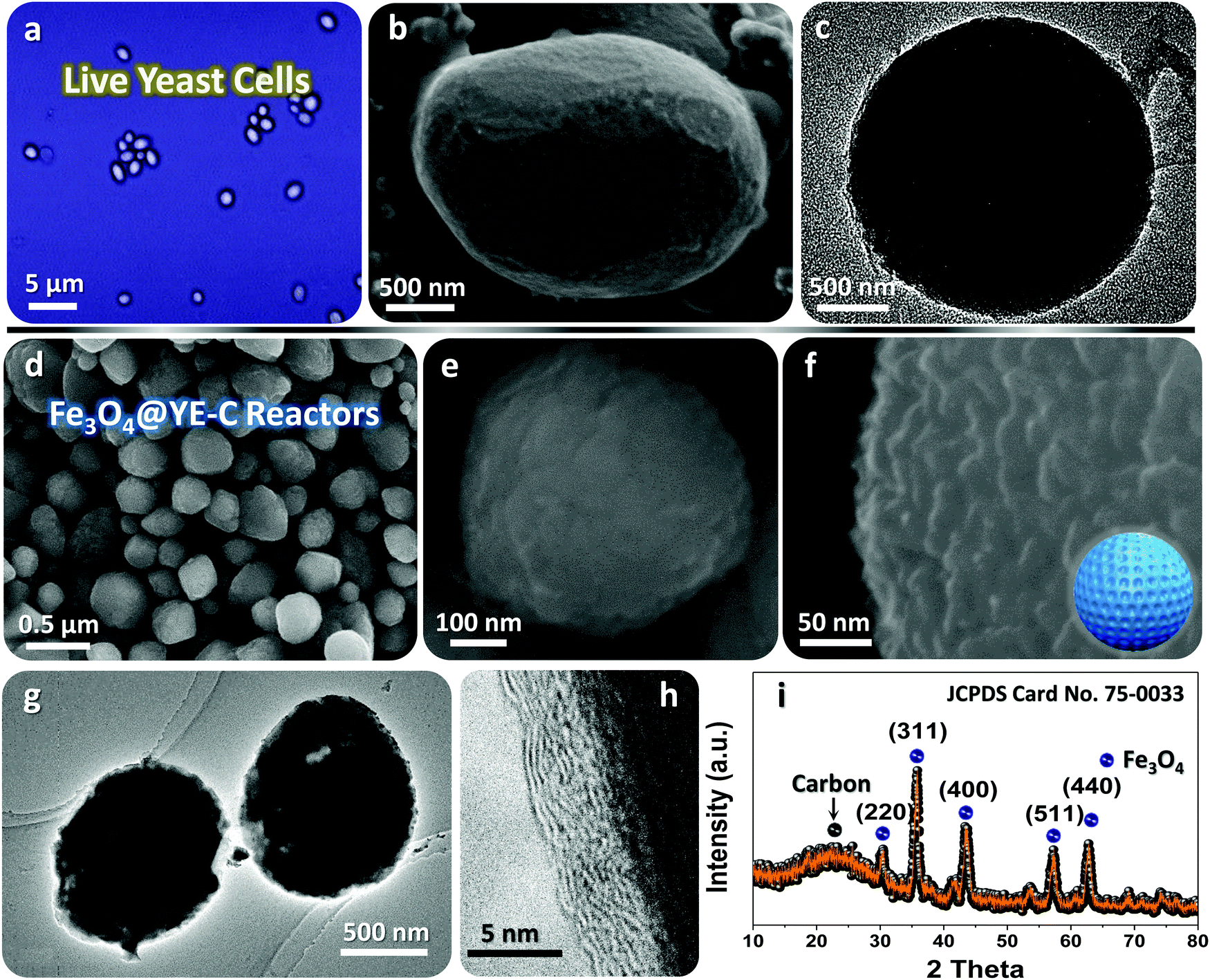 | ||
| Fig. 2 (a) Microscopic, (b) SEM and (c) TEM observations on fresh tiny yeasts. (d–f) SEM and (g and h) TEM observations, as well as (i) XRD pattern of the ultimate Fe3O4@YE-C reactors. | ||
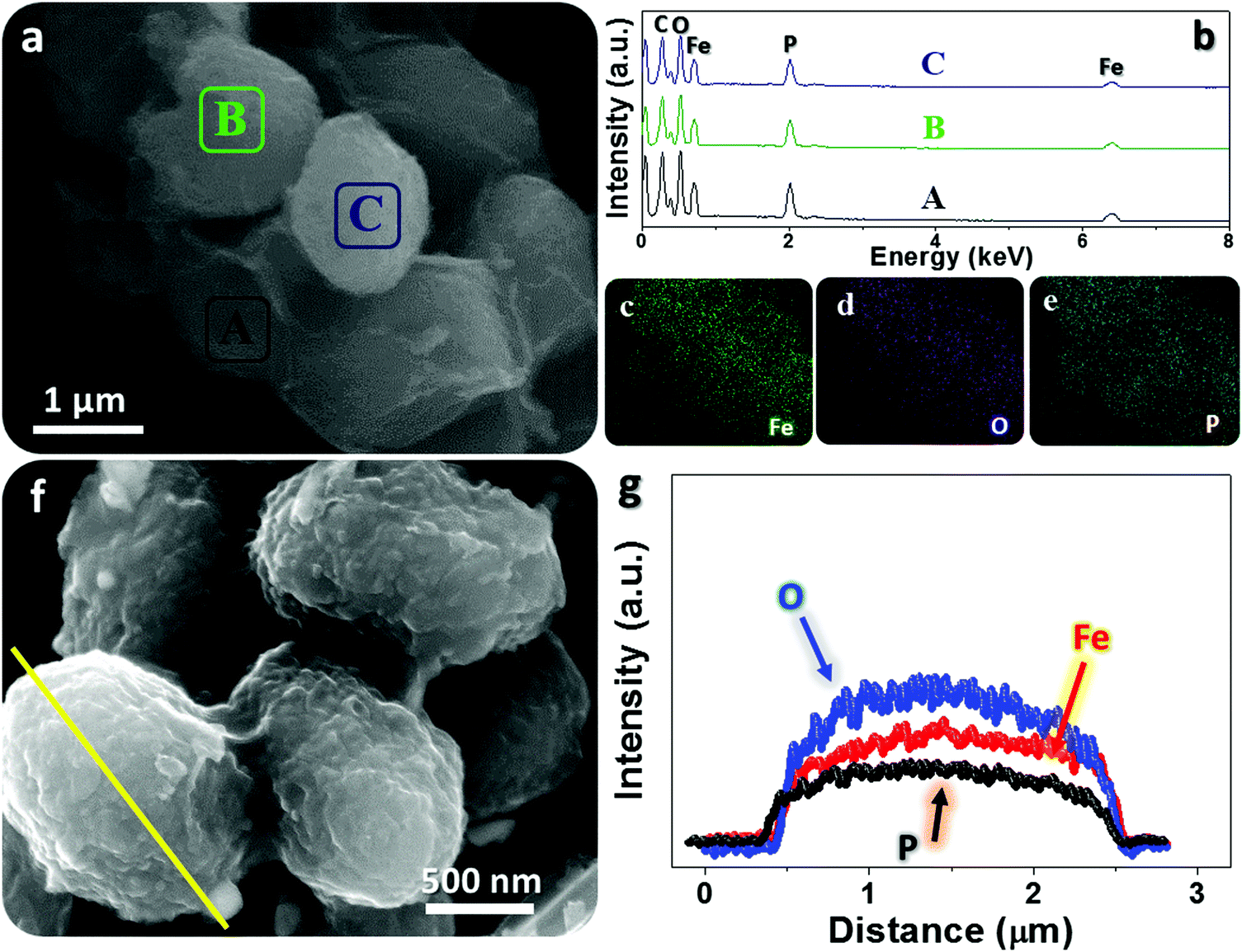 | ||
| Fig. 3 EDS probing toward Fe3O4@YE-C reactors: (a–e) EDS patterns and elemental mapping records for distinct sample units; (f and g) line-scan analysis on a selected particle section. | ||
To clarify the key influences on the engineering beginning with live cells and better control of the evolution procedure, we performed systematic investigations on the relationship between the reaction time and yeast microstate changes (e.g., bioactivity, geometric structure, cell integrity, etc.). Fig. 4a–c displays typical SEM images of samples obtained at specific reaction times. After 30 min soaking in concentrated FeCl3 solution, the basic features are nearly the same as those of pristine cells (see Fig. 2b). Nevertheless, there are salt-like domains emerging on the outer surface of the samples, which may be associated with the peculiar ability of electrostatic adherence for bioactive yeasts.6 Given their intact/smooth surface and unique biological capabilities (e.g., salt tolerance, acid resistance, etc.), we believe most of these fungi may be kept alive at this evolution stage although the ambient environment (pH value: ∼2.1) is truly detrimental and harmful to yeast survival. As time proceeds to 60 min, there are evident structural defects emerging on the involved yeast cells (Fig. 4b); such imperfections may provide convenient channels for Fe3+ diffusion into the inner cell regions. Despite the presence of biochemical reactions, the spherical/elliptical geometric profile of the samples can be still maintained. When the soaking time increases to 90 min, hardly any intact/individual yeasts can be found, except for some cell debris (Fig. 4c). This phenomenon may be fully understandable since biological autolysis would inevitably be launched when cells are immersed in tough salty/acidic solutions for a long time. Thermogravimetric measurements have been made for FeCl3-filled yeast products obtained after the 60 min ion-diffusion and drying procedures (Fig. 4d). By referring to the TG ratio of the remaining Fe2O3 (45.5%) and the slat pyrolysis reaction of 2FeCl3 + 3H2O → Fe2O3 + 6HCl, we could approximatively calculate the mass content of FeCl3 (ratio value: ∼92.4%; note that this is not the actives ratio, and is only used to estimate the salt loaded into yeasts in the soaking process). Moreover, to ensure the salt loading amount at different soak reaction stages, TG analysis on samples made by specific reaction times has been deliberately performed (see the curve in Fig. 4e). The salt loading ratio initially rises slowly (<25%) for times below 45 min, which may be closely related to the specific tolerance ability of fresh yeasts; they are able to hinder entrance of Fe3+ into their inner body. Once the time exceeds 45 min, the ratio value jumps to ∼66.7% (45 min) and then to ∼92.4% (60 min). The sudden changes hint at the gradual invalidation of the cytomembrane, and therefore the vast majority of metallic ions can be filled into the cell body. However, the continual increase in soaking time (≥75 min) would in turn give rise to a decrease in the salt loading amount due to adverse biomass destruction/autolysis, which is in line with our former SEM observations.
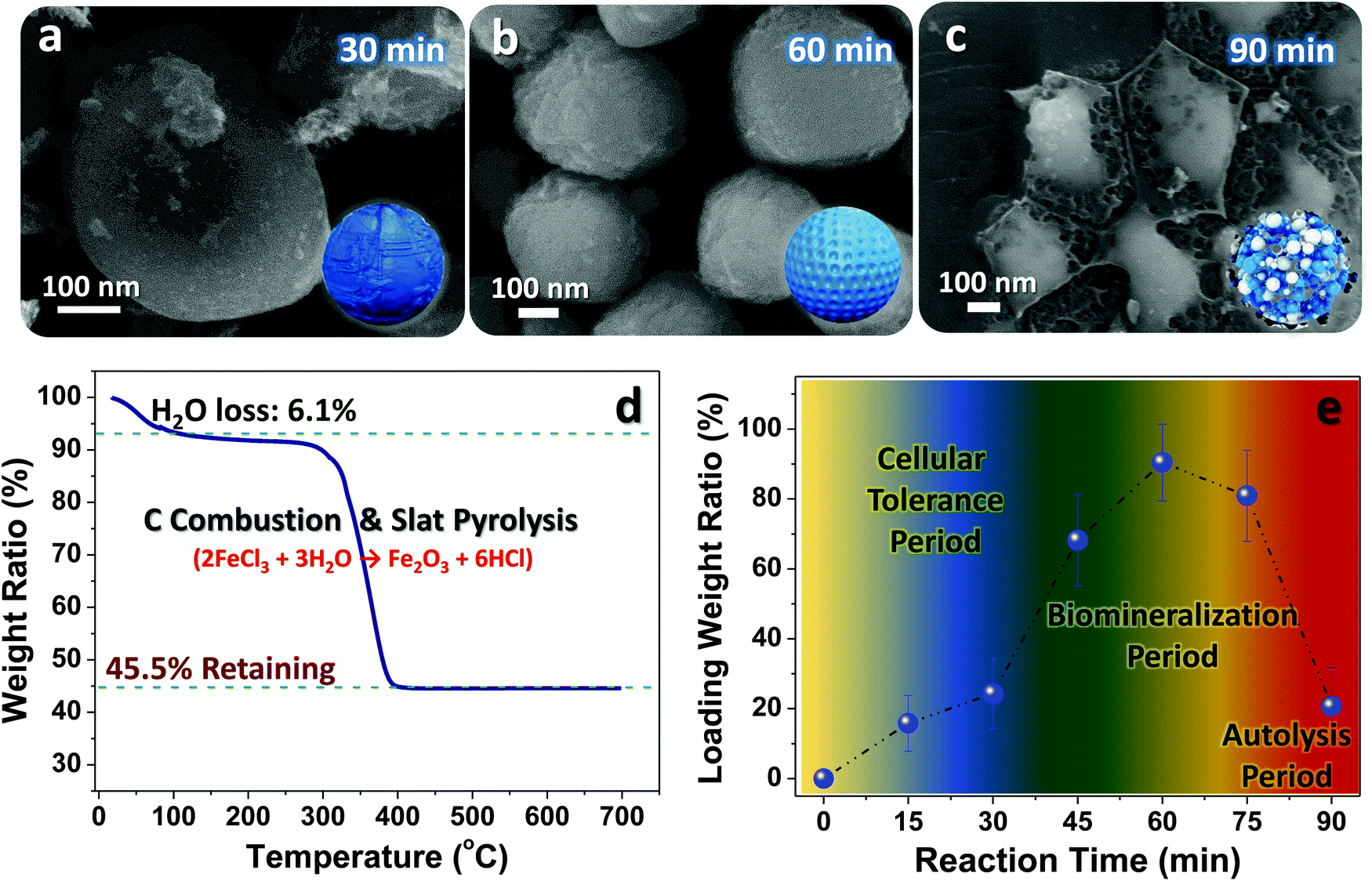 | ||
| Fig. 4 (a–c) SEM monitoring of the morphological changes of Fe3O4@YE-C reactors upon increased reaction time. (d) TG analysis record of FeCl3-filled yeasts made after 60 min soaking reaction and (e) the salt loading ratio calculation for samples at specific reacting stages. | ||
The anodic performance of Fe3O4@YE-C has been evaluated in a three-electrode system by using 3 M KOH as the aqueous electrolyte. To highlight the significant functions of YE-C reactors, anodic testing for Fe3O4@C and bare Fe3O4 nanoparticles is also presented. The electrochemical behaviors of Fe3O4@YE-C are firstly unveiled by cyclic voltammogram (CV) measurement (Fig. 5a). In initial CV scan, pairs of symmetric cathodic/anodic peaks can be clearly seen in a potential range of −1.35 to −0.35 V (vs. Ag/AgCl) at a scan rate of 5 mV s−1 without an extra, time-consuming pre-activation process. This highlights the outstanding electrochemical reversibility and activity of Fe3O4@YE-C. The strong reduction peak at approximately −1.1 V in cathodic scans corresponds to the redox conversion reaction from Fe3+ to Fe2+. Furthermore, Fe2+ is reduced to Fe0 when the potential reaches a more negative position (close to hydrogen evolution). In reverse anodic scans, Fe0 is stepwise oxidized into Fe3+; the weak peak lying at approximately −1.06 V originates from the reversible phase variations from Fe0 to Fe(OH)ads/Fe2+, whereas the one at approximately −0.79 V stems from the Faradaic conversion of Fe2+/Fe3+.24–28Fig. 5b shows CV curves for the Fe3O4@YE-C electrode at scan rates varying from 5 to 40 mV s−1. Upon a gradual rise in scan rate, there is no significant deformation appearing on the CV profiles; in particular, no remarkable cathodic/anodic peak shifts are noticed (e.g., rate: 5 mV s−1, peak potential: −1.1 V; rate: 40 mV s−1, peak potential: −1.16 V), revealing their admirable electrochemical stability and rapid redox reaction kinetics. Fig. 5c displays galvanostatic discharge curves at distinct current densities from ∼1.1 to ∼17.6 A g−1. All discharge curves exhibit a well-defined potential plateau around ∼0.8 V, corresponding well to the oxidation conversion of Fe2+/Fe3+. According to the above discharge records, the stored capacity is calculated and plotted as a function of current rate (Fig. 5d). When operated at ∼1.1 A g−1, such a hybrid electrode can deliver a maximum specific capacity/capacitance of ∼218.2 mA h g−1/∼759.3 F g−1, which is comparable to that of other mainstream anodic counterparts in secondary charge storage devices.29 As is known, stored capacity/capacitance will degrade upon increase of current rates due to unavoidable kinetic restrictions related to both ionic diffusion and electrical transfer. For conventional Fe3O4@C and bare Fe3O4 electrodes, their specific capacity/capacitance reduces sharply to a value of ∼89.1 mA h g−1/∼207 F g−1 and ∼59.7 mA h g−1/∼123 F g−1 when the current rises to ∼32 A g−1, with a capacity retention ratio of ∼24.6% and ∼36.4%, respectively. Our electrodes, however, can retain a value as high as ∼103.0 mA h g−1/∼297 F g−1. This implies that YE-C reactors can indeed support/guarantee the anodic electrical conductivity, and in the meantime supply ample ionic channels for high-power input/output applications, which is confirmed by the low charge transfer resistance (Rct) in the electrochemical impedance (EIS) spectrum (Fig. S3, ESI†). Besides, a long-lasting cyclic test was performed using galvanostatic charge/discharge tests at a constant current rate of ∼1.1 A g−1 (see Fig. 5e). The output capacity of the Fe3O4@YE-C electrode is maintained at ∼100% within the initial 2000 cycles, without any obvious decay, verifying the YE-C function of cyclic stability enhancement. This may be attributed to the fact that such closed reactors can supply protective shells to disperse tiny Fe3O4 nanocrystals and prevent their detrimental aggregation during deep cycling This is fully supported by SEM/TEM observations on cycled Fe3O4@YE-C (see Fig. S4, ESI†), wherein the elliptical profile of each hybrid unit almost stays intact and is clearly distinguished even after a long-time charge/discharge operation. By contrast, bare the Fe3O4 electrode shows dramatic capacity fading, with only ∼80% of the initial capacity retained after a limited period of ∼600 cycles. Although the Fe3O4@C hybrids exhibit far superior cyclic endurance over bare samples, their essential electrochemical pre-activation process (from the 1st to 500th cycle) and undesirable capacitive decline after 1700 cycles still remain problematic for further development. Electrochemical performance comparisons between this work and other classic Fe3O4/C hybrid anodes are present in Table 1, revealing that our Fe3O4@YE-C hybrids exhibit excellent electrode behaviors in charge storage applications.
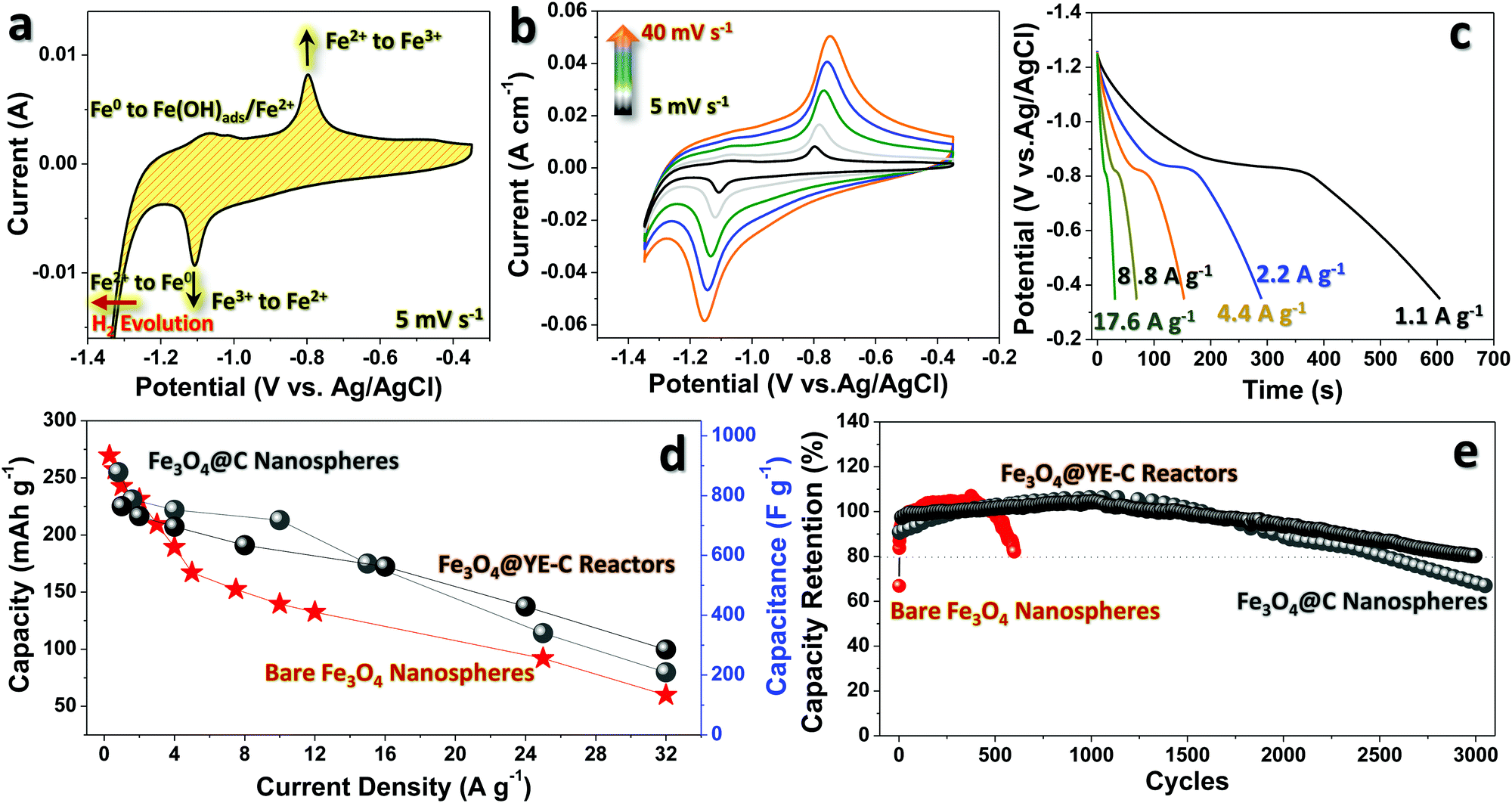 | ||
| Fig. 5 Half-cell electrochemical testing of the Fe3O4@YE-C electrode: (a) a typical CV scan at 5 mV s−1, (b) CV plots at specific scan rates, (c) galvanostatic discharge curves, (d) the plot for the specific capacity as a function of current densities, and (e) the long-term cyclic record. | ||
| Electrode material | Max. C | Rate performance | Cycle performance | Ref. |
|---|---|---|---|---|
| Fe3O4@YE-C | 759.3 F g−1/218.2 mA h g−1 at 1.1 A g−1 | 297 F g−1/103.0 mA h g−1 at 32 A g−1 | ∼98.9% after 2000 cycles at 1.6 A g−1 | This work |
| Fe3O4–carbon arrays | 247.5 mA h g−1 at 2 mV s−1 | 220.43 mA h g−1 at 2 A g−1 | ∼83.9% after 2000 cycles | 30 |
| Fe3O4/CNFs | 135 F g−1 at 10 mV s−1 | 127 F g−1 at 0.42 A g−1 | ∼91% after 1000 cycles | 31 |
| Fe3O4/rGO | 220.1 F g−1 at 0.5 A g−1 | 134.6 F g−1 at 5 A g−1 | ∼99% after 2000 cycles | 32 |
| Porous Fe3O4/carbon composite | 139 F g−1 at 0.5 A g−1 | 74 F g−1 at 5 A g−1 | ∼90% after 2000 cycles | 33 |
| Ultrathin nanoporous Fe3O4–C nanosheets | 163.4 F g−1 at 1 A g−1 | 113 F g−1 at 10 A g−1 | ∼97% after 2000 cycles | 34 |
| Carbon nanotube/Fe3O4 | 117.2 F g−1 at 10 mA cm−2 | 103.6 F g−1 at 40 mA cm−2 | ∼91% after 500 cycles | 35 |
To further evaluate Fe3O4@YE-C electrodes for practical use, a full-cell supercapacitor device has been built by pairing this battery-type anode with the pseudo-capacitive cathode of α-Co(OH)2 NWs (Fe3O4@YE-C(−)//α-Co(OH)2(+)) (basic structural and electrochemical properties for the Co(OH)2 NWs cathode can be found in Fig. S5, ESI†). The schematic for the device configuration and CV testing record are shown in Fig. 6a. Clearly, the CV profile inherits both the anodic and cathodic characteristics, exhibiting a couple of symmetric redox peaks and good reversibility in electrochemical conversions. The major current signal at ∼1.48 V in the anodic scan refers to the generation of CoOOH and Fe2+/Fe0 during the charge process, while the broad signal centered at ∼1.0 V in the cathodic scan is attributed to reverse discharge reactions, wherein Fe2+ or Fe0 species lose electrons and change into Fe3+-based oxides and in the meantime the CoOOH is reduced into Co2+-based hydroxides. The overall reaction can be expressed as follows:
| Fe3O4 + 4Co2+ + 4OH− + 4e− ↔ 3Fe + 4CoOOH |
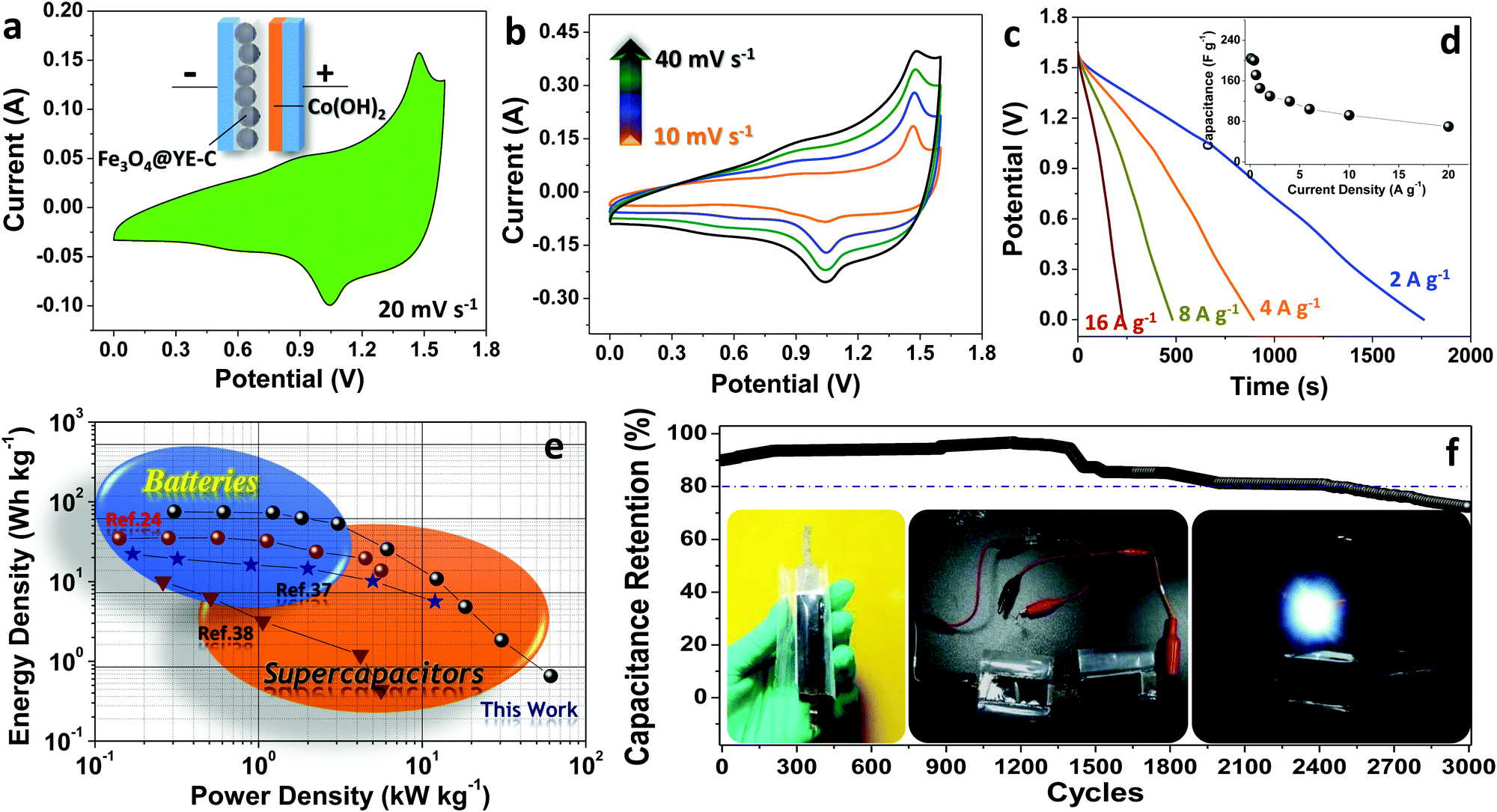 | ||
| Fig. 6 Full-cell electrochemical testing of the Fe3O4@YE-C(−)//α-Co(OH)2(+) device: (a) a typical CV scan at 20 mV s−1, (b) CV plots at specific scan rates, (c) galvanostatic discharge curves, (d) the plot for the specific capacity as a function of current densities, (e) Ragone plot and (f) the long-term cyclic record. | ||
Fig. 5b shows CV curves for Fe3O4@YE-C(−)//α-Co(OH)2(+) devices at scan rates varying from 10 to 40 mV s−1. The absence of any great peak shifts or CV profile distortions verifies their excellent electrochemical reaction kinetics. We further performed galvanostatic testing on the assembled full-cell supercapacitors under different current rates (corresponding discharge profiles are shown in Fig. 6c). The delivered capacitance was calculated/plotted as a function of current rate (Fig. 6d). When running at ∼0.1 A g−1, the full cell can deliver a maximum specific capacitance of ∼231 F g−1. Along with the increase in current densities, the output capacitance is recorded as ∼221 F g−1 (∼0.2 A g−1), ∼209 F g−1 (∼0.4 A g−1), ∼209 F g−1 (∼0.8 A g−1) and ∼203 F g−1 (∼1.6 A g−1), respectively. Even at a large current density of ∼20 A g−1 (∼200 times that of the lowest current rate; the discharge process finishes in a few seconds), the supercapacitors still retain a capacitance as high as ∼83 F g−1. To further show their great potential in practical applications, the Ragone plot of energy density versus power density is presented in Fig. 6e. Clearly, the maximum energy density for Fe3O4@YE-C(−)//α-Co(OH)2(+) devices reaches ∼72 W h kg−1 under a power density of ∼0.31 kW kg−1; even when working at an extremely high-power density of ∼17 kW kg−1, they can still deliver a total energy density of ∼2.6 W h kg−1. Such values are comparable or even surpass those recorded for advanced supercapacitor systems.24,30–38 The long cyclic performance has been examined by galvanostatic charge/discharge tests at ∼1.6 A g−1 (Fig. 6f). After 3000 cycles of fatigue cycling, over ∼70% of the initial capacity is retained, illustrating excellent cyclic stability and endurance. In an aim to prove their potential use, two device units (size: 1 × 3 cm2; total electrode area: ∼6 cm2) have been connected in series to drive a 3 mm diameter white light-emitting-diode (LED; nominal voltage: 2.8 V, rated current: 20 mA; see optical images in the inset of Fig. 6f). The fully charged devices can power the LED indicator brightly for at least hundreds of seconds, suggesting great promise for use in emergency power supply services.
Conclusions
In summary, the approach of directly engineering sustainable charge storage applications starting from live biomass has been demonstrated. By using fresh yeasts as the initial materials, smart pregnant hybrids of Fe3O4@YE-C are evolved via the facile biosynthesis and calcination treatments. Fe3O4 nanoparticles are in situ formed and solidly impregnated in YE-C spheres, which can act as reliable and robust electrochemical reactors for reversible redox conversions. Their construction formation mechanism has been systematically studied by carefully monitoring the microstates of yeast cells upon the varied reaction time. Due to effective combination/synergistic effects between such C reactors and encapsulated Fe3O4 nanoactives, the configured hybrids show excellent anodic behaviors in aqueous electrolytic systems, with a max. specific capacity/capacitance of ∼218.2 mA h g−1/∼759.3 F g−1, admirable rate capability (a capacity/capacitance of ∼103.0 mA h g−1/∼297 F g−1 is retained at ∼32 A g−1), and prolonged cyclic behaviors when compared to the cases for bare Fe3O4 and traditional Fe3O4@C electrodes. A full-cell supercapacitor of Fe3O4@YE-C(−)//α-Co(OH)2(+) is further built to verify the use potential of Fe3O4@YE-C in real applications. The optimized full devices (with max. specific energy and power densities up to ∼72 W h kg−1 and ∼17 kW kg−1) can readily light up the white high-power LED indicators for tens of minutes, exhibiting a great promise as reliable emergency power sources in our daily life. Our work offers an interesting and unconventional way to achieve smart actives@biocarbon configurations for charge storage utilizations. This may open a sustainable and affordable platform to covert the vast of biomaterials/useless household wastes into useful hybrid systems, not merely for electrochemical applications but also for catalyst or biosensor usage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from National Natural Science Foundation of China (11604267, 51802269 and 21773138), Chongqing Natural Science Foundation (cstc2016jcyjA0477 and cstc2018jcyjAX0624) and the Fundamental Research Funds for the Central Universities (XDJK2018C005, SWU115027, and SWU115029). This project is also supported by Program for Innovation Team Building at Institutions of Higher Education in Chongqing (CXTDX201601011, XDJK2017A002).Notes and references
- Q. Ma, Y. Yu, M. Sindoro, A. G. Fane, R. Wang and H. Zhang, Adv. Mater., 2017, 29, 1605361 CrossRef PubMed.
- J. Jiang, J. H. Zhu, W. Ai, Z. X. Fan, X. N. Shen, C. J. Zou, J. P. Liu, H. Zhang and T. Yu, Energy Environ. Sci., 2014, 7, 2670 RSC.
- Z. H. Huang, T. Y. Liu, Y. Song, Y. Li and X. X. Liu, Nanoscale, 2017, 9, 13119 RSC.
- J. J. Liu, Y. F. Deng, X. H. Li and L. F. Wang, ACS Sustainable Chem. Eng., 2016, 4, 177 CrossRef CAS.
- P. Sennu, V. Aravindan, M. Ganesan, Y. G. Lee and Y. S. Lee, ChemSusChem, 2016, 9, 849 CrossRef CAS PubMed.
- S. S. Liu, W. Y. Bian, Z. R. Yang, J. H. Tian, Z. F. Zhou and R. Z. Yang, J. Mater. Chem. A, 2014, 2, 18012 RSC.
- R. Zhang, X. X. Jing, Y. T. Chu, L. Wang, W. J. Kang, D. H. Wei, H. B. Li and S. L. Xiong, J. Mater. Chem. A, 2018, 6, 17730 RSC.
- J. Li, F. R. Qin, L. Y. Zhang, K. Zhang, Q. Li, Y. Q. Lai, Z. A. Zhang and J. Fang, J. Mater. Chem. A, 2014, 2, 13916 RSC.
- L. Y. Zhang, Y. Y. Wang, B. Peng, W. T. Yu, H. Y. Wang, T. Wang, B. W. Deng, L. Y. Chai, K. Zhang and J. X. Wang, Green Chem., 2014, 16, 3926 RSC.
- X. Q. Zhang, Y. Zhong, X. H. Xia, Y. Xia, D. H. Wang, C. A. Zhou, W. J. Tang, X. L. Wang, J. B. Wu and J. P. Tu, ACS Appl. Mater. Interfaces, 2018, 10, 13598 CrossRef CAS PubMed.
- Y. Li, G. Wang, T. Wei, Z. Fan and P. Yan, Nano Energy, 2016, 19, 165–175 CrossRef CAS.
- J. H. Zhu, S. Y. Liu, Y. N. Liu, T. Meng, L. Ma, H. Zhang, M. Q. Kuang and J. Jiang, ACS Sustainable Chem. Eng., 2018, 6, 13662–13669 CrossRef CAS.
- J. Jiang, H. Zhang, J. H. Zhu, L. P. Li, Y. N. Liu, T. Meng, L. Ma, M. W. Xu, J. P. Liu and C. M. Li, ACS Appl. Mater. Interfaces, 2018, 10, 24157–24163 CrossRef CAS PubMed.
- J. Jiang, J. Zhu, W. Ai, X. Wang, Y. Wang, C. Zou, W. Huang and T. Yu, Nat. Commun., 2015, 6, 8622 CrossRef CAS PubMed.
- D. Xie, X. H. Xia, W. J. Tang, Y. Zhong, Y. D. Wang, D. H. Wang, X. L. Wang and J. P. Tu, J. Mater. Chem. A, 2017, 5, 7578–7585 RSC.
- Y. Zhong, X. Xia, S. Deng, J. Zhan, R. Fang, Y. Xia, X. Wang, Q. Zhang and J. P. Tu, Adv. Energy Mater., 2018, 8, 1701110 CrossRef.
- X. D. Zhang, X. G. Zhang, W. He, C. Y. Sun, J. Y. Ma, J. L. Yuan and X. Y. Du, Colloids Surf., B, 2013, 103, 114–120 CrossRef CAS PubMed.
- J. Zhu, L. Li, Z. Xiong, Y. Hu and J. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 269 CrossRef CAS.
- J. Jiang, Y. N. Liu, L. P. Li, J. H. Zhu, M. W. Xu and C. M. Li, ACS Sustainable Chem. Eng., 2018, 6, 757–765 CrossRef CAS.
- C. D. Wang, M. H. Lan, Y. Zhang, H. D. Bian, M. F. Yuen, K. Ostrikov, J. J. Jiang, W. J. Zhang, Y. Y. Li and J. Lu, Green Chem., 2016, 18, 3029 RSC.
- H. Lu and X. S. Zhao, Sustainable Energy Fuels, 2017, 1, 1265 RSC.
- J. Wang, P. Nie, B. Ding, S. Y. Dong, X. D. Hao, H. Dou and X. G. Zhang, J. Mater. Chem. A, 2017, 5, 2411 RSC.
- H. D. Espinosa, J. E. Rim, F. Barthelat and M. J. Buehler, Prog. Mater. Sci., 2009, 54, 1059 CrossRef CAS.
- J. Jiang, L. P. Li, M. W. Xu, J. H. Zhu and C. M. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3874–3882 CrossRef CAS PubMed.
- C. Guan, J. L. Liu, Y. D. Wang, L. Mao, Z. X. Fan, Z. X. Shen, H. Zhang and J. Wang, ACS Nano, 2015, 9, 5198–5207 CrossRef CAS PubMed.
- R. Z. Li, Y. Wang, C. Zhou, C. Wang, X. Ba, Y. Li, X. Huang and J. Liu, Adv. Funct. Mater., 2015, 25, 5384–5394 CrossRef CAS.
- S. Sun, T. Zhai, C. L. Liang, S. V. Savilov and H. Xia, Nano Energy, 2018, 45, 390–397 CrossRef CAS.
- J. Q. Liu, M. B. Zheng, X. Q. Shi, H. B. Zeng and H. Xia, Adv. Funct. Mater., 2016, 26, 919–930 CrossRef CAS.
- Q. Y. Xia, M. Xu, H. Xia and J. P. Xie, ChemNanoMat, 2016, 2, 588–600 CrossRef CAS.
- R. Z. Li, Y. M. Wang, C. Zhou, C. Wang, X. Ba, Y. Y. Li, X. T. Huang and J. P. Liu, Adv. Funct. Mater., 2015, 25, 5384–5394 CrossRef CAS.
- J. B. Mu, B. Chen, Z. C. Guo, M. Y. Zhang, Z. Y. Zhang, P. Zhang, C. L. Shao and Y. C. Liu, Nanoscale, 2011, 3, 5034–5040 RSC.
- Q. H. Wang, L. F. Jiao, H. M. Du, Y. J. Wang and H. T. Yuan, J. Power Sources, 2014, 245, 101–106 CrossRef CAS.
- W. J. Meng, W. Chen, L. Zhao, Y. Huang, M. S. Zhu, Y. Huang, Y. Q. Fu, F. X. Geng, J. Yu, X. F. Chen and C. Y. Zhi, Nano Energy, 2014, 8, 133–140 CrossRef CAS.
- D. Q. Liu, X. Wang, X. B. Wang, W. Tian, J. W. Liu, C. Y. Zhi, D. Y. He, Y. Bando and D. Golberg, J. Mater. Chem. A, 2013, 1, 1952–1955 RSC.
- D. H. Guan, Z. Gao, W. L. Yang, J. Wang, Y. Yuan, B. Wang, M. L. Zhang and L. H. Liu, Mater. Sci. Eng., B, 2013, 178, 736–743 CrossRef CAS.
- R. Z. Li, X. Ba, H. F. Zhang, P. Xu, Y. Y. Li, C. W. Cheng and J. P. Liu, Adv. Funct. Mater., 2018, 1800497 CrossRef.
- J. Jiang, L. P. Li, Y. N. Liu, S. Y. Liu, M. W. Xu and J. H. Zhu, Nanotechnology, 2017, 28, 145402 CrossRef PubMed.
- X. Geng, L. Li and F. Li, Electrochim. Acta, 2015, 168, 25 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00019d |
| ‡ J. Jiang and S. Liu contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |