
Hierarchical CoFe-layered double hydroxide and g-C3N4 heterostructures with enhanced bifunctional photo/electrocatalytic activity towards overall water splitting†
Muhammad
Arif
ab,
Ghulam
Yasin
 ac,
Muhammad
Shakeel
a,
Muhammad Asim
Mushtaq
ab,
Wen
Ye
b,
Xiaoyu
Fang
b,
Shengfu
Ji
*a and
Dongpeng
Yan
*ab
ac,
Muhammad
Shakeel
a,
Muhammad Asim
Mushtaq
ab,
Wen
Ye
b,
Xiaoyu
Fang
b,
Shengfu
Ji
*a and
Dongpeng
Yan
*ab
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: yandongpeng001@163.com; jisf@mail.buct.edu.cn
bKey Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China
cBUCT-CWRU International Joint Laboratory, State Key Laboratory of Organic–Inorganic Composites, Center for Soft Matter Science and Engineering, College of Energy, Beijing University of Chemical Technology, Beijing, China
First published on 28th January 2019
Abstract
To achieve sustainable and clean energy for the hydrogen economy, developing efficient earth-abundant and non-noble transition metal photo/electrocatalysts toward overall water splitting is highly desirable. In this work, layered double hydroxide (LDH)@g-C3N4 composites with hierarchical flower-like micro/nanosheets and a high surface area have been synthesized by a solvothermal method. HRTEM images exhibit that the surface of the g-C3N4 nanosheets is highly orientated with the main exposure of the (002) plane. Compared with pristine CoFe-LDH, the hierarchical nanocomposite presents an excellent and stable elecrocatalytic performance in 1.0 M KOH, with a small Tafel slope of 58 mV dec−1 and an overpotential of about 275 mV at a current density of 10 mA cm−2. Simultaneously, CoFe-LDH@g-C3N4 exhibits an exceptional performance for the HER in 1.0 M KOH electrolyte, with an overpotential of 417 mV at a current density of 10 mA cm−2 and a small Tafel slope of 77 mV dec−1. Therefore, this work not only accomplishes improved catalytic activity of CoFe-LDH by the introduction of g-C3N4 nanosheets, but also provides an insight into the correlation between hierarchical flower-like morphologies and photo/electrochemical catalytic activity for overall water splitting.
The simultaneous production of oxygen (O2) and hydrogen (H2) from photo/electrochemical water-splitting provides one of the most promising approaches to develop low-cost, environmentally friendly and sustainable energy conversion/storage.1–6 However, the commercial applications of photo/electrochemical water splitting are usually limited because the anodic oxygen evolution reaction (OER) and the cathodic hydrogen evolution reaction (HER) are greatly uphill with large overpotentials. Electrocatalysts are critically necessary to minimize the energy barrier and enhance the kinetics of both OER and HER to ensure the overall process is more energy-efficient.7–15 To date, Ru/Ir and Pt based precious metals are the state-of-the-art photo/electrocatalysts for OER and HER, respectively, but their relatively low durability, high cost and scarcity usually limit their large-scale utilization for water splitting. A tremendous amount of research has therefore been conducted to develop low-cost, highly active and durable photo/electrocatalysts with the aim of substituting noble metal-based catalysts.16–24
Recently, photo/electrocatalysts with tunable hierarchical micro/nanostructures have gained significant attention, due to their high surface area, more active sites and proficient light harvesting for enhancing photo/electrocatalytic activity in overall water splitting. Moreover, various micro/nanostructures greatly influence the formation of heterojunctions and the tuning of the band gap.25–32 Non-noble transition metal-based systems, such as single-metal or mixed-metal oxides and hydro(-oxy)oxides, are propitious for maintaining water oxidation with low overpotential and good durability under alkaline conditions. Hence, the construction of new non-noble metal-based semiconductor photo/electrocatalysts with hierarchical morphologies could be an effective way to enhance efficiency in the water splitting process.33–37
As a large type of two-dimensional (2D) semiconductor material, layered double hydroxides (LDHs) have been used as photo/electrocatalysts in water splitting due to their remarkable activity, low cost and easy preparation. Generally, LDHs are represented by the formula [MII1−xMIIIx(OH)2]z+(An−)z/n·[yH2O], in which MII, MIII and An− represent divalent and trivalent metallic cations and interlayer guest anions compensating for metal cations, respectively.1,2,5,12,38–47 Simultaneously, another low-cost and metal-free 2D polymeric system, graphitic carbon nitride (g-C3N4), has also been evaluated, mostly for electrochemical and photoelectrochemical water splitting due to its engaging electronic structure, visible-light absorption, high stability (thermal and chemical) and environmental friendliness.11,48–56 Furthermore, g-C3N4 composites have also been investigated for broad energy conversion techniques (such as CO2 reduction to hydrocarbon, environmental purification and the photodegradation of organic pollutants).57–63 To date, low surface area, weak electrical conductivity and easy recombination of electron–hole pairs are major disadvantages associated with g-C3N4.11,64–66 To overcome these aforementioned problems, tremendous efforts (doping with metals, coupling with other semiconductors, and/or copolymerization) have been made to improve the catalytic activity of g-C3N4.49,65–69 In this sense, the heterojunctions formed between g-C3N4 and LDH semiconductors could be a splendid strategy for boosting their photo/electrocatalytic properties since their similar 2D-layered characteristics are helpful to their heterostructural coupling, full charge separation and electron transfer. Additionally, the hierarchical morphologies and tunable chemical compositions of LDHs can be elegantly modified, which facilities an enhancement in electrical conductivity and charge separation. Recently, Shakeel et al. reported Ni–Mn-LDH/g-C3N4 composite for electrocatalytic water splitting with an overpotential of 316 mV to achieve a current density of 10 mA cm−2; Tanmay et al. reported that Co0.4Fe0.6-LDH/g-CNx required overpotentials of 280 mV and 377 mV to acquire current densities 10 mA cm−2 and 100 mA cm−2. However, there is still a gap for researchers to develop new catalysts with unique architectures and special morphologies towards high catalytic activity at low overpotential. From an elemental point of view, it is also documented that Co2+/Co3+, Fe2+/Fe3+ and Co–N are the active sites for boosting electrochemical overall water splitting.11,52,64,70–74 Additionally, the effect of visible light on electrocatalytic activities remains a subject of scientific interest.70
To combine the advantages of Co-based 2D LDHs and g-C3N4, in this work, hierarchical CoFe-LDH/g-C3N4 composites have been chosen as model systems, which are synthesized via a simple one-step and easily scalable in situ solvothermal method. Their electrocatalytic and photo/electrocatalytic activities are systematically investigated. The hierarchical features of a CoFe-LDH@g-C3N4 heterostructure could provide various reflection sites and cavities for the absorption of light and consequently exhibit excellent catalytic activity for overall water splitting. The electrical conductivity of a CoFe-LDH@g-C3N4 composite also increases due to electronic interactions and the unique morphology between CoFe-LDH and g-C3N4 layers at 10 wt% loading of g-C3N4 content. Thus, the CoFe-LDH@g-C3N4 composite exhibits improved photo/electrocatalytic OER and HER activity and durability compared with its individual components. The photocurrent density of pure CoFe-LDH and hierarchical CoFe-LDH@g-C3N4 composite are 2.1 and 3.2 times that of g-C3N4, respectively. Furthermore, a current density of 10 mA cm−2 is achieved by applying just 1.55 V, while a current density of 50 mA cm−2 is reached by applying just 1.76 V across a two-electrode electrolyzer with the hierarchical CoFe-LDH@g-C3N4 composite acting as both anode and cathode during the overall water splitting process. Therefore, this work not only affirms the active role of the hierarchical heterostructure of the CoFe-LDH@g-C3N4 composite as co-catalysts in photo/electrochemical water splitting but also provides further insights into similar 2D semiconductors for renewable energy applications.
Experimental details
Materials
Chemicals Co(NO3)2·6H2O, Fe(NO3)3·9H2O, KOH, anhydrous N,N-dimethylformamide (DMF), C2H5OH, and Al2O3 powder were purchased from Sinopharm Chemical Reagent (Beijing Co. Ltd) All reagents were analytical grade and were used without further purification. Deionized water was used throughout the experiments.Preparation of g-C3N4
Bulk g-C3N4 was synthesized by the thermal treatment of dicyandiamide in a muffle furnace. Typically, 5.0 g of dicyandiamide was ground into powder and heated at 550 °C for 4 hours at a heating rate of 2.5 °C min−1. After cooling, the yellow solid was collected and ground into powder until the yellow color of bulk g-C3N4 changed to light yellow. g-C3N4 nanosheets were synthesized by further thermal treatment of bulk g-C3N4. Light yellow powder was calcined at 600 °C for 2 hours at a heating rate of 5.0 °C min−1, and was further modified by liquid exfoliation. The g-C3N4 sample was ultrasonicated in water for 10 hours and then separated by centrifugation. The resulting powder was washed three times with pure water and ethanol, collected and then dried at 60 °C overnight.Fabrication of the hierarchical flower-like CoFe-LDH@g-C3N4 composite
Typically, a certain amount of the g-C3N4 nanosheets was dispersed in 6 mL of anhydrous N,N-dimethylformamide (DMF) assisted by ultrasonication for more than 1 hour. The hierarchical flower-like CoFe-LDH nanosheets were synthesized by a facile solvothermal method. After that, 600 μL of 0.2 M Co(NO3)2·6H2O and 200 μL of 0.2 M Fe(NO3)3·9H2O aqueous solutions were added into an ultrasonicated mixture containing g-C3N4. For the adsorption of metal cations through the electrostatic interaction on the surface of g-C3N4, the mixture of CoFe-LDH and g-C3N4 was treated ultrasonically for at least 1 hour. For a typical run, Co(NO3)2·6H2O and Fe(NO3)3·9H2O with a Co/Fe molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was optimized for photo/electrocatalytic activity. Firstly, the mixture was strenuously stirred at 85 °C for 4 hours. Then, the product was obtained and dispersed in a mixture of DMF (6 mL) and water (12 mL). After being stirred vigorously for 4 hours, the obtained mixture was transferred to a 40 mL Teflon-lined stainless steel autoclave. The reaction mixture in the autoclave was treated at 120 °C and maintained for 24 hours. The resulting product was separated by centrifugation and washed thoroughly with deionized water and ethanol more than three times. At the end, the obtained product was dried in a vacuum oven at 50 °C for one night. Pure CoFe-LDH was synthesized by the same procedure as aforementioned but without adding g-C3N4 nanosheets and with no ultrasonication throughout the reaction.
:1 was optimized for photo/electrocatalytic activity. Firstly, the mixture was strenuously stirred at 85 °C for 4 hours. Then, the product was obtained and dispersed in a mixture of DMF (6 mL) and water (12 mL). After being stirred vigorously for 4 hours, the obtained mixture was transferred to a 40 mL Teflon-lined stainless steel autoclave. The reaction mixture in the autoclave was treated at 120 °C and maintained for 24 hours. The resulting product was separated by centrifugation and washed thoroughly with deionized water and ethanol more than three times. At the end, the obtained product was dried in a vacuum oven at 50 °C for one night. Pure CoFe-LDH was synthesized by the same procedure as aforementioned but without adding g-C3N4 nanosheets and with no ultrasonication throughout the reaction.
Synthesis of the CoFe-LDH and CoFe-LDH@g-C3N4 nanosheets
As control samples, CoFe-LDH nanosheets were synthesized by a facile co-precipitation and hydrothermal method. For a typical process, Co(NO3)2·6H2O and Fe(NO3)3·9H2O were dissolved in deionized water, forming a clear solution with a Co/Fe molar ratio of 3, keeping the total metal-ion concentration at 1.2 mol L−1. Two different solutions including NaOH (1.92 M) and Na2CO3 (0.80 M) were also prepared with equal volumes of basic and salt solutions, respectively. After that, both solutions were mixed together and added into a beaker containing the metal salt solution under vigorous stirring. The resulting slurry was kept in an oven at 80 °C for 48 h. After cooling naturally, the precipitate was washed several times with deionized water and ethanol by centrifugation, and dried at room temperature. For comparison, a CoFe-LDH@g-C3N4 composite was also prepared by the same procedure with the addition of different amounts of g-C3N4.Characterizations
Powder X-ray diffraction (XRD) patterns of all of the samples were conducted using graphite-filtered Cu Kα radiation operating at 40 kV and 30 mA and λ = 0.15418 nm (Shimadzu XRD-6000 diffractometer). X-ray photoelectron spectrometry (XPS) was recorded using Al Kα radiation (Thermo VG ESCALAB MK II). The positions of all BEs were calibrated using the C 1s line at 284.8 eV. Scanning electron microscopy with an accelerating voltage of 20 kV (SEM, Zeiss SUPRA 55) was applied for detailed morphology analyses. Transmission electron microscope (TEM) and EDS mapping were taken using a microscope (JEOL JEM-2010F) combined with an EDX (OxfordX-MaxN 80-TLE) spectroscope. Photoluminescence (PL) experiments were performed on an Edinburgh FLS980 fluorescence spectrometer.Electrode preparation
Typically, 5 mg of the as-synthesized catalyst was dispersed in 1000 μL of deionized H2O; 250 μL of ethyl alcohol and 15 μL of Nafion solution were ultrasonically treated for at least 1 hour to form a homogeneous mixture. Then, 10 μL of the mixture was drop-caste onto the surface of a glassy carbon (GC) electrode by micro-pipette and the GC allowed to dry by evaporation of the solvent at room temperature.Electrochemical and photoelectrochemical (PEC) measurements
All electrochemical and photoelectrochemical (PEC) measurements for OER and HER were conducted on an electrochemical workstation (CHI 660 C, CH Instrument Co. USA) using 1 M KOH solution as an electrolyte. All tests were carried out in a three-electrode electrochemical cell, i.e. Ag/AgCl (reference electrode) with saturated KCl filling solution, Pt wire (counter electrode) and a GC electrode with a diameter of 3 mm (working electrode). For activation, the GC electrode was polished with alumina powder on a nylon polishing pad. Then, the electrode was completely rinsed with deionized H2O and cleaned in acetone by ultrasonication for 5 s. For 1 M KOH as an electrolyte, the applied potential changed from E vs. Ag/AgCl to overpotential and reversible hydrogen electrode (RHE), E vs. RHE = E vs. Ag/AgCl + 1.009 V, and the overpotential (η) for OER was η = E vs. RHE − 1.23 V = E vs. Ag/AgCl − 0.221 V. Cyclic voltammograms (CVs) were carried out between 0.2 and 0.8 V vs. Ag/AgCl at a scan rate of 100 mV s−1 for 30 cycles to activate the electrode electrochemically in 1 M KOH. LSV was conducted between 0.2 and 0.8 V vs. Ag/AgCl at a scan rate of 10 mV s−1; Tafel slopes and polarization curves were calculated from the LSV curves. Furthermore, an electrochemically active surface area (ECSA) was measured from the capacitive current associated with double-layer charging from CVs at different scan rates. For this purpose, small potential windows (0.2–0.3 V) of CVs were measured versus Ag/AgCl at different scan rates (10, 20, 40, 60, 80 and 100 mV s−1). The double-layer capacitance (Cdl) was investigated by plotting the ΔJ = (Ja − Jc) at 0.25 V versus Ag/AgCl against the scan rate and the linear slope was found to be twice that of Cdl. Tafel slopes were calculated from the LSV curves by plotting log(J) on the x-axis and η on the y-axis. In a similar cell setup (three-electrode configuration), electrochemical impedance spectroscopy (EIS) evaluation was carried out to determine the electrical conductivity of the as-synthesized catalysts. The frequency range for EIS was from 100 kHz to 0.10 Hz with a modulation voltage amplitude of 5 mV at 0.5 V across Ag/AgCl potential. A durability (time and current density curves at a constant potential of 0.5 V across Ag/AgCl) test for the hierarchical CoFe-LDH@g-C3N4 composite was conducted on a three-electrode photo/electrochemical cell configuration using 1 M KOH solution as an electrolyte. PEC measurements were carried out using a CHI660C electrochemical workstation (CH Instruments Co. USA) with a three-electrode system, in which Pt wire, Ag/AgCl and Ni foam were used as the counter, reference and working electrodes, respectively. The working electrode was irradiated under a 300 W xenon arc lamp with an AM 1.5G filter (illumination intensity: 100 mW cm−2) using 1 M KOH aqueous solution as an electrolyte.Photoluminescence spectroscopy (PL)
PL spectroscopy was conducted to examine the charge transfer behavior between g-C3N4 and CoFe-LDH.Overall water splitting
The overall water splitting reaction was performed on a two-electrode system, in which hierarchical CoFe-LDH@g-C3N4 acts as anode and cathode in 1.0 M KOH electrolyte. The two electrodes were placed ∼2 cm apart from each other to avoid mixing of gases (H2 and O2) generated during the overall water splitting reaction, and the assembly design was opened to the air to release the gases during water splitting. The as-synthesized homogeneous ink of the hierarchical CoFe-LDH@g-C3N4 composite was assembled by drop-casting onto 1 × 1 cm2 Ni foam and the LSV curves were measured at a scan rate of 10 mV s−1 with iR-compensation.Calculation method
The TOF value is determined from the equation: TOF = j × S/(4F × m). Here, j (mA cm−2) is current density at a given η (in our case at 350 mV), S (0.07 cm2) is the surface area of the GC electrode, the number 4 means 4 electrons per mole of O2, F is Faraday's constant (96485.3 C mol−1), and m is the number of moles of metal deposited on the GC electrode. In this case, we suppose that all the metal sites are actively participating in the PEC reaction.Results and discussion
The crystallographic structure and phase purity of the as-prepared samples were firstly evaluated by powder X-ray diffraction (XRD), as shown in Fig. 1. For g-C3N4, two peaks appeared at ca. 13.0° and 27.8°, in which the sharp one at 27.8° confirms the high degree of graphitization of the as-prepared composite. The weaker graphitization intensity of the product at 13.0° is due to an in-planar structural packing motif at the (100) plane. Stacking of the conjugated aromatic systems in the product leads to a strong peak at 27.8° that is assigned to the (002) plane in the (00l) direction. For CoFe-LDH, diffraction peaks at 11.4°, 23.2°, 34.0°, 38.4°, 46.1°, 59.3° and 60.4° can be ascribed to the (003), (006), (012), (015), (018), (110) and (113) diffraction planes, respectively, for typical 2D LDH systems. The XRD pattern of hierarchical CoFe-LDH@g-C3N4 confirms the presence of both g-C3N4 and CoFe-LDH individual compounds in the resulting composite. Additionally, the peak intensity of g-C3N4 at 27.8° increases without disturbing the peak positions of CoFe-LDH by increasing the weight percentage loading of g-C3N4 (3–15 wt%) on CoFe-LDH. This means that the formation of g-C3N4 nanosheets does not hinder the crystal growth of CoFe-LDH. Hence it could be inferred that the crystal structures of g-C3N4 and CoFe-LDH remain unaffected during the formation of the CoFe-LDH@g-C3N4 heterostructure.64,70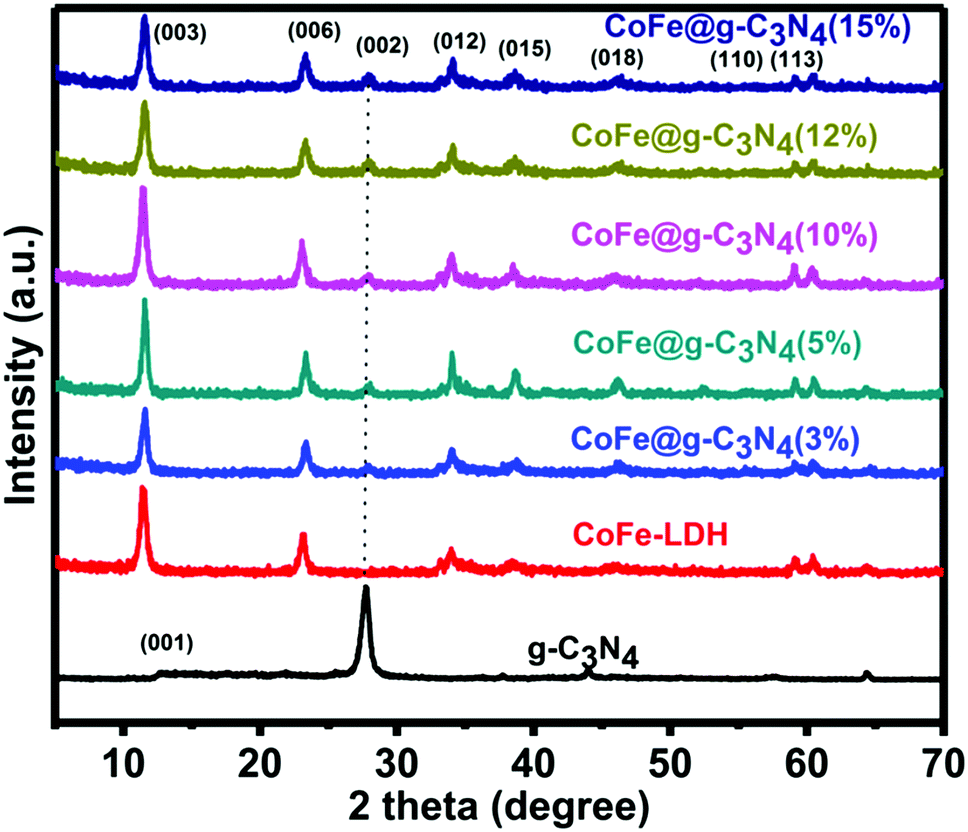 | ||
| Fig. 1 XRD patterns of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 composite materials. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were performed to evaluate the morphological and structural features of the g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 systems. For pure g-C3N4, SEM exhibits curved and irregular 2D small nanosheets, which form the aggregation structure (Fig. 2a). While for pure CoFe-LDH, the SEM images exhibit a hierarchical flower-like morphology with an amount of cavities, with a thickness of 10–15 nm and length of 100–150 nm (Fig. 2b and Fig. S1a, b, ESI†). This hierarchical flower-like morphology is totally different from the traditional hexagonal micro/nanosheet morphology of LDHs, which may be related to the formation of multiple nucleation centers during the solvothermal process (Scheme 1).
 | ||
| Fig. 2 SEM images of g-C3N4 (a), CoFe-LDH (b) and CoFe-LDH@g-C3N4 composites (c and d); EDS mapping of CoFe-LDH@g-C3N4 composite (e). | ||
 | ||
| Scheme 1 Schematic illustration of the fabrication of a CoFe-LDH@g-C3N4 hybrid semiconductor for photoelectrochemical overall water splitting. | ||
Upon formation of CoFe-LDH@g-C3N4 (Fig. 2c and d), the morphologies of the composites vary from those of their individual components. Taking CoFe-LDH@g-C3N4 (10%) as an example, the g-C3N4 nanosheet arrays are elegantly dispersed within the hierarchical flower-like CoFe-LDH. Flower and petal-like nanosheets of CoFe-LDH become more corrugated and curlier after combination with g-C3N4 during the in situ solvothermal process (Fig. S1c and d, ESI†). Such a CoFe-LDH@g-C3N4 architecture may lead to a greater electrochemically active surface area due to corrugation of the flower-like structure, optimal pore-size and reduced ion transport distance. Energy dispersive spectra (EDS) of CoFe-LDH@g-C3N4 (10%) obviously prove the overlapping of all corresponding elements of Co, Fe, C, N and O, authenticating the uniform dispersion of CoFe-LDH and g-C3N4 in the composite (Fig. 2e and Fig. S2, ESI†). Furthermore, TEM was characterized to evaluate the detailed nanostructures of pure CoFe-LDH and CoFe-LDH@g-C3N4 composites. The CoFe-LDH exhibits obvious petal-like structures composed of ultrathin nanosheets and some cavities (Fig. 3a and Fig. S3a, b, ESI†). CoFe-LDH@g-C3N4 exhibits a high dispersion of g-C3N4, and the corrugated LDHs nanosheets are interlaced at the edges of the nanocomposites (Fig. 3b and c), which is consistent with the SEM images. The lattice fringe spacing values of 0.20, 0.23 and 0.32 nm can be attributed to the interplanar spacings of the (018), (015) and (002) planes for the CoFe-LDH and g-C3N4 nanosheets, respectively (Fig. 3d–f), which are consistent with their XRD patterns. The criss-crossing between the CoFe-LDH and g-C3N4 lattices also indicates the formation of heterostructures (Fig. S3c and d, ESI†). The selected-area electron diffraction (SAED) pattern provides an indication of the polycrystalline state and homogeneous distribution of g-C3N4 on the CoFe-LDH layers through the appearance of diffraction rings (Fig. 3d inset). Such interfacial characteristics may facilitate an increase in ECSA and the separation of the interfacial charge between CoFe-LDH and g-C3N4.
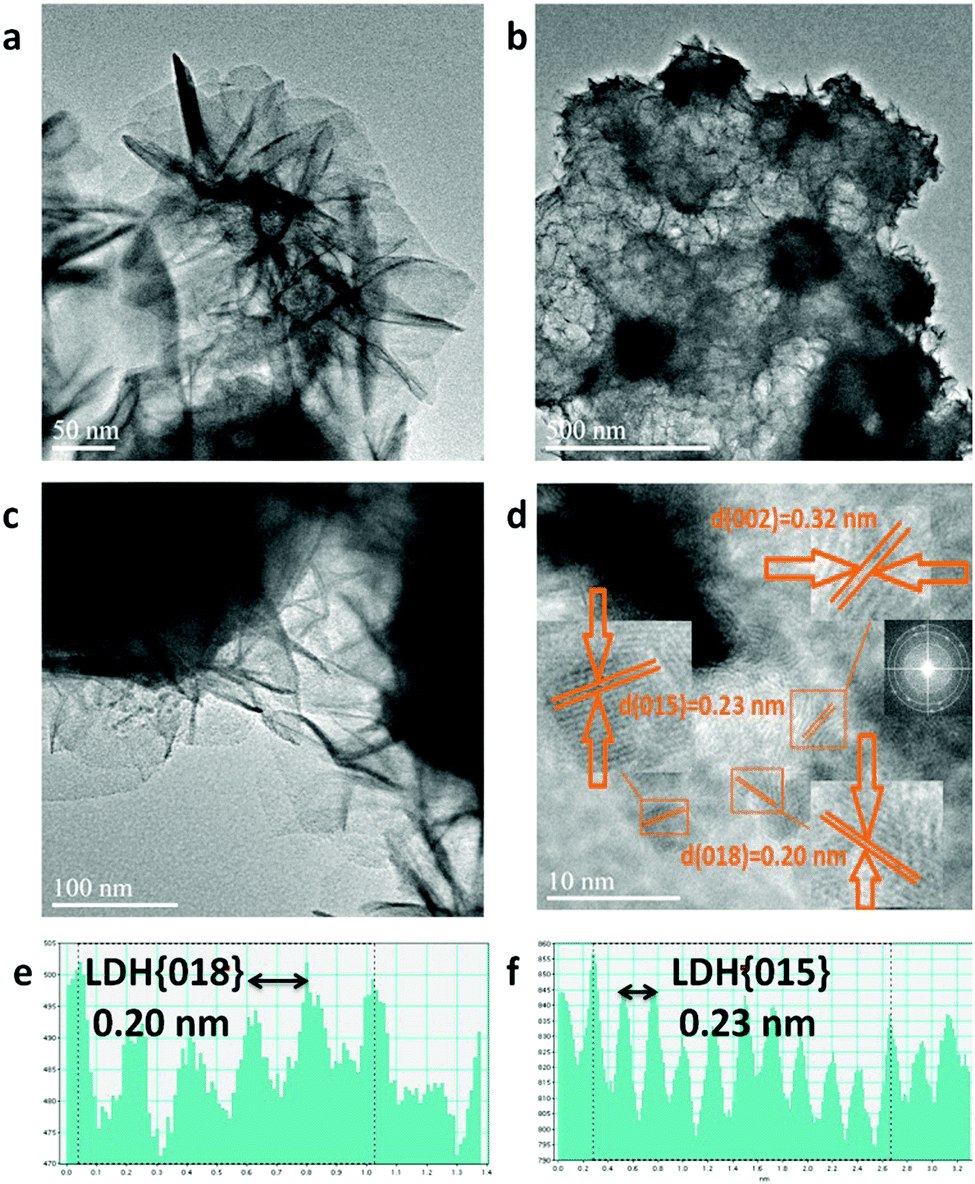 | ||
| Fig. 3 TEM images of CoFe-LDH (a) CoFe-LDH@g-C3N4 composite (b–d) (inset in figure d: photography of CoFe-LDH@g-C3N4 hybrid and SAED pattern) and the profile of the distance between lattice fringes of hierarchical LDH in the CoFe-LDH@g-C3N4 hybrid (e and f). | ||
The optical properties of the g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 composites were evaluated using diffuse reflectance UV-Vis spectroscopy (Fig. S7, ESI†). The spectra with absorbance less than 500 nm or higher than 500 nm can be attributed to the metal-to-metal charge transfer (MMCT) and metal-to-ligand charge transfer (MLCT), respectively.75–77 MLCT arises from the 2p orbital of oxygen to the 3d orbitals of Co2+/Fe3+ ions (i.e. O2p/Co3d-t2g, O2p/Fe3d-t2g).78,79 The g-C3N4 exhibits an intrinsic semiconductor-like absorption spectrum with a band edge of 450 nm (blue region of the visible spectrum) and can be assigned to the n–π* direct transition of lone pairs on the edge of the N atoms.70,80 Thus, the modification of CoFe-LDH with g-C3N4 comprehensively changes the optical properties in the resultant hierarchical CoFe-LDH@g-C3N4 heterostructure. As displayed in Fig. S7 (ESI†), all the modified composites (CoFe-LDH@g-C3N4) with different weight loadings of g-C3N4 exhibit obvious absorption over the whole visible range of wavelengths. With an increase in loading of g-C3N4, an optimal change in the absorption edges is observed in the spectra, and this change is attributed to the strong interaction between g-C3N4 nanosheets and CoFe-LDH. An extremely high visible light absorption ability of the CoFe-LDH@g-C3N4 heterostructures is attributed to the dual roles of the delocalized conjugated structure for g-C3N4 and CoFe-LDH (1s/π* and 1s/o′* transitions, and LMCT and d–d transitions, respectively).64 The optimized optical absorption spectrum of the CoFe-LDH@g-C3N4 composite occurs at a content of 10 wt% of g-C3N4. The maximum absorption of the composites is highly related to the unique morphology, exposed active sites and high surface area. However, further increasing the content of g-C3N4 (>10 wt %) may lead to a shielding effect of the active sites, disturbing the layered structure and blocking the active sites on the surface of CoFe-LDH, resulting in decreased electronic excitation under irradiation. The band gap energy of a crystalline semiconductor can be calculated from the following expression:
| (αhν)1/n = A(hν − Eg) |
The separation and transfer efficiencies of the photogenerated hole and electron pairs in various semiconductor materials are evaluated by PL. The PL spectra of g-C3N4, CoFe-LDH, and CoFe-LDH@g-C3N4 (10%) heterostructures are shown in Fig. S4 (ESI†). The main peak of g-C3N4 is centered at approximately 440–450 nm, which is associated with the typical photoemission of g-C3N4, close to the band-gap energy of 2.7 eV and is totally consistent with the UV-Vis and Eg results. The sharp decrease in the PL intensity for the CoFe-LDH@g-C3N4 (10%) composite proves that the recombination of photogenerated electron–hole pairs is obviously suppressed with the maximum photoelectrocatalytic activity towards overall water splitting upon the formation of the CoFe-LDH@g-C3N4 (10%) composite due to the synergistic coupling of CoFe-LDH and g-C3N4 nanosheets. This is correlated with the dynamic and fast charge transfer from g-C3N4 to CoFe-LDH, which can be favorable for enhancing the PEC water oxidation reaction. In addition, the diffuse reflectance UV-Vis results of CoFe-LDH@g-C3N4 (10%) support the PL spectra. These results suggest that the strong electronic coupling of the g-C3N4 nanosheets with the flower-like CoFe-LDH matrix facilitates the efficient movement of photogenerated excitons of the g-C3N4 layers during energy transfer. Additionally, the hierarchical flower-like surface of CoFe-LDH could act as trapping sites for photogenerated electrons, which may reduce the rate of electron–hole recombination for enhanced photoelectrocatalytic activity during water splitting.
XPS studies were conducted to analyze the oxidation state and chemical bonding of the as-prepared heterostructures (Fig. 4). According to full-scan XPS spectra, composite materials include all C, N, O, Co, and Fe elements from g-C3N4 and CoFe-LDH (Fig. 4a). The fitting analysis of the C 1s XPS spectra of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 is shown in Fig. 4b, which exhibits the presence of graphitic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (∼284.82 eV), and C–NC bonds (∼288.22 eV) for both g-C3N4 and CoFe-LDH@g-C3N4, while extra peaks at ∼283.77 (C–H), 285.12 (CN), 287.27 (carbonyl carbon, CO), 287.87 (N–CO) and 288.52 eV (carboxyl groups, O–CO) appear only for CoFe-LDH@g-C3N4. Furthermore, the N 1s XPS spectra of CoFe-LDH@g-C3N4 and g-C3N4 (Fig. 4c) present three almost similar peaks with binding energies of 400.87, 398.92 and 398.42 eV, corresponding to quaternary-N, N–C and pyridinic-N, respectively. In the N 1s spectra of hierarchical CoFe-LDH@g-C3N4, the composite exhibits one additional peak to be found at ∼399.42 eV (Co–N) relative to pure g-C3N4. These nitrogen centers are favorable to OER, and in particular the appearance of the Co–N bond will benefit a speedy redox process.11,81–83 The presence of additional bonds (C–O, CO, O–CO, and Co–N) and an additional peak around 781.8 eV signify that CoFe-LDH@g-C3N4 is not a simple physical mixture, but has a strong chemical interaction between the heterostructures. Such a structure could accelerate the electronic coupling between two components and generate extra active sites (i.e. interfaces) for OER and HER during the overall water splitting process.11,16,66,67 The XPS spectrum also proves the presence of Fe in the as-prepared composite material, as shown in Fig. 4d. The Fe species in the CoFe-LDH@g-C3N4 composite can be deconvoluted into five peaks, which are dominated by the Fe3+ oxidation state. The peak at 711.2 eV assigned to Fe 2p3/2 and the peak at 725.1 eV assigned to Fe 2p1/2 are consistent with the occurrence of Fe(OH)3 units in CoFe-LDH. Furthermore, the fitting result of the Co 2p peak indicates two oxidation states recognized as Co2+ and Co3+ (Fig. 4e). In the Co 2p XPS spectra, the peaks at 780.17 and 796.6 eV are assigned to Co3+ and the peaks at 786.2 and 802.9 eV are attributed to Co2+. The electrocatalytic activities of hierarchical CoFe-LDH@g-C3N4 were explored for both OER and HER studies with a three-electrode electrochemical cell in 1 M KOH solution (Fig. 5). Fig. 5a exhibits the polarization curve after iR correction (scan rate: 10 mV s−1). The linear sweep voltammetry (LSV) curves reveal overpotentials (to achieve a current density of 10 mA cm−2) of 525, 314, 268 and 275 mV for g-C3N4, CoFe-LDH, RuO2 and CoFe-LDH@g-C3N4 (10%), respectively (Fig. S5, ESI†). To achieve a current density of 60 mA cm−2, g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%) show overpotentials of 548, 408 and 322 mV, respectively. These observations affirm that the individual g-C3N4 and CoFe-LDH deliver very high overpotential and lower current densities, but when they are combined into hierarchical heterostructures, the catalytic activity is tremendously enhanced. Moreover, the photoelectrochemical performances for water splitting were also studied. Typically, the anodic and cathodic reactions (OER and HER) are accompanied by the transfer of a majority of carriers (holes and electrons, respectively). The photoexcitation process changes the amount of carriers at the interface and the current density is therefore enhanced by illumination. Hence, the photoelectrochemical results show a decrease in overpotential. In the presence of light, the hierarchical CoFe-LDH@g-C3N4 composite exhibits an obviously decreased overpotential at 270 mV (Fig. 5a). Furthermore, an obvious decrease in overpotential from 380 mV to 346 mV is also noted to achieve a current density of 100 mA cm−2 for CoFe-LDH@g-C3N4 (10%) under visible light illumination, confirming the high-efficiency PEC OER effect (Fig. S6, ESI†). The optimized photoelectrochemical activity of the composites occurs at a content of 10 wt%. In our opinion, the photo/electrochemical performance of the composites is highly related to their unique morphology, having good light harvesting capability, with a high surface area and exposed active sites to facilitate charge transportation in the heterostructure. The turnover frequency (TOF) is also an important kinetic parameter for OER, illustrating the intrinsic properties of the electrocatalytic activity. Here, it is assumed that all metal sites in the CoFe-LDH@g-C3N4 composite are electrochemically active in the water oxidation reaction, and their turnover frequencies at an overpotential of 350 mV can be calculated. The results give values of TOF = 0.32 s−1, 0.237 s−1, 0.213 s−1, 0.123 s−1, 0.094 s−1 and 0.046 s−1, corresponding to CoFe-LDH@g-C3N4 (10%) with light, without light, CoFe-LDH@g-C3N4 (12%), CoFe-LDH@g-C3N4 (5%), CoFe-LDH@g-C3N4 (3%) and CoFe-LDH, respectively. It is worth mentioning that the above TOFs are obviously underestimated because some metal sites may be electrochemically inaccessible.
C (∼284.82 eV), and C–NC bonds (∼288.22 eV) for both g-C3N4 and CoFe-LDH@g-C3N4, while extra peaks at ∼283.77 (C–H), 285.12 (CN), 287.27 (carbonyl carbon, CO), 287.87 (N–CO) and 288.52 eV (carboxyl groups, O–CO) appear only for CoFe-LDH@g-C3N4. Furthermore, the N 1s XPS spectra of CoFe-LDH@g-C3N4 and g-C3N4 (Fig. 4c) present three almost similar peaks with binding energies of 400.87, 398.92 and 398.42 eV, corresponding to quaternary-N, N–C and pyridinic-N, respectively. In the N 1s spectra of hierarchical CoFe-LDH@g-C3N4, the composite exhibits one additional peak to be found at ∼399.42 eV (Co–N) relative to pure g-C3N4. These nitrogen centers are favorable to OER, and in particular the appearance of the Co–N bond will benefit a speedy redox process.11,81–83 The presence of additional bonds (C–O, CO, O–CO, and Co–N) and an additional peak around 781.8 eV signify that CoFe-LDH@g-C3N4 is not a simple physical mixture, but has a strong chemical interaction between the heterostructures. Such a structure could accelerate the electronic coupling between two components and generate extra active sites (i.e. interfaces) for OER and HER during the overall water splitting process.11,16,66,67 The XPS spectrum also proves the presence of Fe in the as-prepared composite material, as shown in Fig. 4d. The Fe species in the CoFe-LDH@g-C3N4 composite can be deconvoluted into five peaks, which are dominated by the Fe3+ oxidation state. The peak at 711.2 eV assigned to Fe 2p3/2 and the peak at 725.1 eV assigned to Fe 2p1/2 are consistent with the occurrence of Fe(OH)3 units in CoFe-LDH. Furthermore, the fitting result of the Co 2p peak indicates two oxidation states recognized as Co2+ and Co3+ (Fig. 4e). In the Co 2p XPS spectra, the peaks at 780.17 and 796.6 eV are assigned to Co3+ and the peaks at 786.2 and 802.9 eV are attributed to Co2+. The electrocatalytic activities of hierarchical CoFe-LDH@g-C3N4 were explored for both OER and HER studies with a three-electrode electrochemical cell in 1 M KOH solution (Fig. 5). Fig. 5a exhibits the polarization curve after iR correction (scan rate: 10 mV s−1). The linear sweep voltammetry (LSV) curves reveal overpotentials (to achieve a current density of 10 mA cm−2) of 525, 314, 268 and 275 mV for g-C3N4, CoFe-LDH, RuO2 and CoFe-LDH@g-C3N4 (10%), respectively (Fig. S5, ESI†). To achieve a current density of 60 mA cm−2, g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%) show overpotentials of 548, 408 and 322 mV, respectively. These observations affirm that the individual g-C3N4 and CoFe-LDH deliver very high overpotential and lower current densities, but when they are combined into hierarchical heterostructures, the catalytic activity is tremendously enhanced. Moreover, the photoelectrochemical performances for water splitting were also studied. Typically, the anodic and cathodic reactions (OER and HER) are accompanied by the transfer of a majority of carriers (holes and electrons, respectively). The photoexcitation process changes the amount of carriers at the interface and the current density is therefore enhanced by illumination. Hence, the photoelectrochemical results show a decrease in overpotential. In the presence of light, the hierarchical CoFe-LDH@g-C3N4 composite exhibits an obviously decreased overpotential at 270 mV (Fig. 5a). Furthermore, an obvious decrease in overpotential from 380 mV to 346 mV is also noted to achieve a current density of 100 mA cm−2 for CoFe-LDH@g-C3N4 (10%) under visible light illumination, confirming the high-efficiency PEC OER effect (Fig. S6, ESI†). The optimized photoelectrochemical activity of the composites occurs at a content of 10 wt%. In our opinion, the photo/electrochemical performance of the composites is highly related to their unique morphology, having good light harvesting capability, with a high surface area and exposed active sites to facilitate charge transportation in the heterostructure. The turnover frequency (TOF) is also an important kinetic parameter for OER, illustrating the intrinsic properties of the electrocatalytic activity. Here, it is assumed that all metal sites in the CoFe-LDH@g-C3N4 composite are electrochemically active in the water oxidation reaction, and their turnover frequencies at an overpotential of 350 mV can be calculated. The results give values of TOF = 0.32 s−1, 0.237 s−1, 0.213 s−1, 0.123 s−1, 0.094 s−1 and 0.046 s−1, corresponding to CoFe-LDH@g-C3N4 (10%) with light, without light, CoFe-LDH@g-C3N4 (12%), CoFe-LDH@g-C3N4 (5%), CoFe-LDH@g-C3N4 (3%) and CoFe-LDH, respectively. It is worth mentioning that the above TOFs are obviously underestimated because some metal sites may be electrochemically inaccessible.
 | ||
| Fig. 4 (a) Full-scan XPS spectra of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%); (b) C 1s of g-C3N4, LDH and CoFe-LDH@g-C3N4; (c) N 1s of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%); (d) Fe 2p spectra of CoFe-LDH and CoFe-LDH@g-C3N4 (10%); (e) Co 2p spectra of Co. | ||
 | ||
| Fig. 5 (a) Linear scan voltammogram (LSV) OER curves of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (with different wt%). (b) Corresponding Tafel slopes for OER. (c) Chronoamperometry of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%) in light and dark applying E = +0.20 V vs. Ag/AgCl for 400 s in 1 M KOH solution. (d) EIS of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%). (e) LSV HER curves of Pt/C, g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%). (f) Corresponding Tafel slopes for HER. | ||
The superior performance of CoFe-LDH@g-C3N4 (10 wt%) can be associated with its hierarchical flower-like morphology and abundant active sites (such as quaternary N, Co–N, pyridinic N, Co2+ and Co3+, as confirmed by XPS). However, a further increase in the content of g-C3N4 (>10 wt%) may lead to the shielding effect of active sites and electron channels in LDHs, as well as the penetration of free electrolyte, which are not beneficial to an enhancement in catalytic activities. Remarkably, the low overpotential to achieve a current density (10 mA cm−2) value for CoFe-LDH@g-C3N4 is comparable to some other state-of-the-art OER catalysts (Table S1, ESI†). The efficient photo/electrocatalytic activity of the hierarchical heterostructure is further confirmed by the low Tafel slope (Fig. 5b). The CoFe-LDH@g-C3N4 composite shows a much lower Tafel slope value (58 mV dec−1) relative to its individual components. In contrast, the pristine g-C3N4, CoFe-LDH and RuO2 exhibit higher Tafel slope values of 114, 82, and 78 mV dec−1, respectively. It is believed that a lower Tafel slope correlates with a speedy catalytic reaction, and thus encourages high OER activity. Under visible light, the Tafel slope of hierarchical CoFe-LDH@g-C3N4 further decreases to 51 mV dec−1, which confirms the higher PEC catalytic activity, and is consistent with the trend in LSV. To further investigate the conductivity, electrochemical impedance spectroscopies (EIS) of hierarchical CoFe-LDH@g-C3N4, CoFe-LDH and g-C3N4 were performed in a three-electrode system. Values of charge transfer resistance (Rct) and solution resistance (Rs) were measured and compared according to the corresponding equivalent circuit values from the Nyquist plots. Both Rs and Rct are resistance across the electrode/electrolyte interface and CPE is the constant phase component (inset in Fig. 5d). Various semi-circles in the Nyquist plots in the high-frequency range can be correlated with the Rct of the as-synthesized electrocatalysts. The diameter of the semicircle illustrates the charge transfer resistance (Rct) and a smaller value of Rct signifies high conductivity as well as faster charge-transfer ability. Therefore, the EIS results (Fig. 5d) prove that the hierarchical CoFe-LDH@g-C3N4 shows a much lower charge transfer resistance (97.02 Ω) relative to CoFe-LDH (157.0 Ω) or g-C3N4 (498.6 Ω), symbolic of its miraculous electron-transfer behavior during the electrochemical reaction. Under visible light irradiation, the EIS of the hierarchical CoFe-LDH@g-C3N4 composite decreases from 97.02 Ω to 76.00 Ω, confirming its high PEC catalytic activity. It is known that typical LDHs undergo low conductivity, and the enhanced electronic coupling in the hierarchical CoFe-LDH@g-C3N4 composite would accelerate the charge transfer and electron conductivity, which agrees well with the LSV and Tafel slope results (Fig. 5b). As shown above, the Tafel slope and overpotential are simultaneously decreased under visible light irradiation. The hierarchical CoFe-LDH@g-C3N4 (10%) heterostructures could play a vital role in the excellent electrochemical activities due to the synergistic effects between the flower-like CoFe-LDH and g-C3N4. To detect the possible PEC mechanism, the transient photocurrent responses of g-C3N4, CoFe-LDH and the hierarchical CoFe-LDH@g-C3N4 composite were also investigated. Fig. 5c exhibits the photocurrent curves of g-C3N4, CoFe-LDH and hierarchical CoFe-LDH@g-C3N4 for certain on–off cycles under visible light. The photocurrent density will go down immediately once the light is turned off, while the photocurrent density will return to a high constant value when the light is turned on. The photocurrent density of CoFe-LDH@g-C3N4 (10%) is calculated by subtracting the dark current from the total current. The photocurrent density of CoFe-LDH@g-C3N4 is 0.196 mA cm−2, which is 2.0 and 3.2 times higher than those of CoFe-LDH and g-C3N4, respectively. The results reveal that the hierarchical CoFe-LDH@g-C3N4 composite shows a higher photocurrent response than its individual parts, indicating that the hierarchical composites dramatically suppress electron–hole recombination and enhance the separation of interfacial charge transfer between CoFe-LDH and g-C3N4 under visible light, which favors an improvement in PEC activity in overall water splitting.
Next, the HER catalytic capabilities of g-C3N4, commercial Pt/C, CoFe-LDH and hierarchical CoFe-LDH@g-C3N4 catalysts were further detected in 1 M basic solution for the probability of overall water splitting (Fig. 5e). The current density (−10 mA cm−2) of hierarchical CoFe-LDH@g-C3N4 appears at −417 mV (η10 = −417 mV). In contrast, CoFe-LDH and g-C3N4 require η10 of −538 mV and −670 mV. To reach a current density of −50 mA cm−2, CoFe-LDH@g-C3N4, CoFe-LDH and g-C3N4 require η50 of −472 mV, −592 mV and >−700 mV, respectively. The overpotential of hierarchical CoFe-LDH@g-C3N4 (η50 = −472 mV) is very close to that of Pt/C (η50 = −416 mV). The HER catalytic kinetics for all the synthesized samples were also evaluated by the Tafel plots, as shown in Fig. 5f. It is believed that there are two mechanisms ((1) Volmer–Tafel or (2) Volmer–Heyrovsky) involving the HER process under basic conditions.23,27 Both mechanisms involve the adsorption of a water molecule, the dissociation of adsorbed water to an adsorbed H atom and OH−, desorption of OH−, and the transformation of adsorbed H into H2, as shown below:
| H2O + e− → Hads + OH−(Volmer); |
| Hads + Hads → H2(Tafel); |
| H2O + e− → Hads + OH−(Volmer); |
| Hads + H2O + e− → H2 + OH−(Heyrovsky) |
The Tafel slope of hierarchical CoFe-LDH@g-C3N4 is 71 mV dec−1, which is smaller than that of CoFe-LDH (95 mV dec−1), g-C3N4 (134 mV dec−1), or Pt/C (136 mV dec−1), demonstrating that hierarchical CoFe-LDH@g-C3N4 has more efficient electrocatalytic kinetics for HER. To determine whether the Tafel, Heyrovsky, or Volmer component is the rate-limiting step, slopes of 30, 40, or 120 mV dec−1 are inspected, respectively. As shown in Fig. 5f, the Tafel value of the hierarchical CoFe-LDH@g-C3N4 composite is 71 mV dec−1, which suggests that the HER over the hierarchical CoFe-LDH@g-C3N4 composite progresses by a Volmer–Heyrovsky mechanism and that the Heyrovsky reaction is the rate-limiting step. The electrochemically active surface area (ECSA) is an immensely vital parameter for catalysts used in the overall water splitting reaction, as it is admitted that an increase in ECSA generally leads to an enhancement in the photo/electrocatalytic activity of the as-prepared catalyst. To gain insight into the improved photo/electrochemical performance of hierarchical CoFe-LDH@g-C3N4 in comparison with the controls, the ECSA were calculated and predicted based on the electrochemical double-layer capacitance (Cdl) derived from different CV curves at different scan rates (Fig. 6). It is obvious that the Cdl value of hierarchical CoFe-LDH@g-C3N4 reaches 36.9 mF cm−2, 2.17 times and 126.3 times higher than those of CoFe-LDH (17 mF cm−2) and g-C3N4 (0.292 g-C3N4), respectively. The higher ECSA indicates that the 2D hierarchically porous configuration could promote gas penetration and release during overall water splitting. Such an improved ECSA at the same mass loading will lead to the exposure of more active sites and maintain an adequate electrochemical reaction between hierarchical CoFe-LDH@g-C3N4 and electrolyte ions, thus delivering high photo/electrocatalytic activity.
 | ||
| Fig. 6 Typical cyclic voltammetry curves (a–c) of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%) electrodes in 1 M KOH with different scan rates; (d) charging current density differences (ΔJ = Ja − Jc) of g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 (10%). | ||
To detect the advantages of the hierarchical flower-like structure of LDH, we also prepared CoFe-LDH@g-C3N4 using a combined co-precipitation and hydrothermal method for comparative analysis. The PXRD patterns, SEM images and electrochemical properties of the as-synthesized g-C3N4, CoFe-LDH and CoFe-LDH@g-C3N4 composites are discussed and displayed in Fig. S8–S10 (ESI†), respectively. Moreover, for additional comparative analysis, a physical mixture (CoFe-LDH and g-C3N4 physically mixed together) of CoFe-LDH and g-C3N4 was also prepared. SEM images and electrochemical properties of the as-prepared physical mixture are discussed and shown in Fig. S11 and S12 (ESI†). The results show that the hierarchical flower-like structure presents a higher PEC electrocatalytical performance than the sample with the other morphology or a purely physical mixture.
The overall splitting of water was performed in a two-electrode system in which hierarchical CoFe-LDH@g-C3N4 acts as both the anode and the cathode in 1 M basic solution (Fig. 7). The LSV curve for overall water splitting shows that it only requires an applied potential of ca. 1.55 V to achieve a current density of 10 mA cm−2 for the hierarchical CoFe-LDH@g-C3N4 electrocatalyst. To deliver a current density of 100 mA cm−2, hierarchical CoFe-LDH@g-C3N4 only requires a cell voltage of ca. 1.82 V for the overall water splitting process. Such a low potential of the CoFe-LDH@g-C3N4 composite for overall water splitting is remarkable and comparable to recently reported bifunctional electrocatalysts (Table S2, ESI†).
 | ||
| Fig. 7 Overall water splitting using CoFe-LDH@g-C3N4 (10%) as OER and HER catalyst. | ||
The magnificent photo/electrocatalytic performance for overall water splitting of the hierarchical flower-like CoFe-LDH@g-C3N4 composite might be attributed to its unique hierarchical flower-like structure and ultrahigh active surface area, as discussed above. Stability is a very important parameter to evaluate the electrocatalytic properties of the as-prepared catalyst for practical application. A stability test was performed with and without light irradiation by continuous galvanostatic measurement for 12 hours.
Impressively, the hierarchical CoFe-LDH@g-C3N4 composite electrocatalyst exhibits good stability and durability in 1 M KOH basic solution in the presence and absence of light irradiation, as shown in Fig. 8. Furthermore, by applying 0.5 V potential, the current density maintained at ∼16.5 mA cm−2 for a period of 12 hours is an indication of good durability. The high stability performance illustrates that the hierarchical flower-like CoFe-LDH@g-C3N4 composite structure is stable and suitable as a practical electrocatalyst for water splitting.
 | ||
| Fig. 8 Durability test of CoFe-LDH@g-C3N4 (10%) in 1 M KOH solution (time-dependent current density curve under constant potential of 0.5 V versus Ag/AgCl) with and without a light source. | ||
Conclusions
In summary, we report for the first time, the synthesis of hierarchical flower-like CoFe-LDH@g-C3N4 as a 2D porous composite through a facile solvothermal method. Our study reveals the opportunities provided by these unique hierarchical structures to function as extremely active and highly stable micro/nanostructures for high-performance bifunctional OER and HER. The hierarchical CoFe-LDH@g-C3N4 electrode shows magnificent activity toward overall water splitting, with 10 mA cm−2 current density achieved by only 1.55 V and with 100 mA cm−2 current density by 1.82 V across a two-electrode system. The magnificent HER and OER activities are associated with the strong electronic interactions between combined components, as proved by the XPS (such as graphitic N and Co–N), SEM and HRTEM evaluations, which show the increased charge transfer and conductivity. The CoFe-LDH@g-C3N4 composite exhibits enhanced current density at a lower overpotential and high stability, indicating its potential practical applications for the overall water splitting process. Therefore, this work provides an efficient way to achieve a new type of hierarchical flower-like CoFe-LDH@g-C3N4 system towards full water splitting in energy applications, and supplies a detailed understanding of the PEC OER and HER processes of the 2D hybrid semiconductors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21473013, 21771021, and 21822501), the Beijing Nova Program (xx2018115), the Fundamental Research Funds for the Central Universities, and Analytical and Measurements Fund of Beijing Normal University.Notes and references
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef.
- J. Liu, J. Wang, B. Zhang, Y. Ruan, L. Lv, X. Ji, K. Xu, L. Miao and J. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 15364–15372 CrossRef CAS PubMed.
- Y. Xue, Z. Zuo, Y. Li, H. Liu and Y. Li, Small, 2017, 13, 1700936 CrossRef PubMed.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- R. Gao, H. Zhang and D. Yan, Nano Energy, 2017, 31, 90–95 CrossRef CAS.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef CAS PubMed.
- D. Kong, H. Wang, Z. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed.
- Q. Liu, J. Tian, W. Cui, P. Jiang, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., 2014, 53, 6710–6714 CrossRef CAS PubMed.
- J. Yu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 11229–11235 RSC.
- M. Tahir, N. Mahmood, L. Pan, Z.-F. Huang, Z. Lv, J. Zhang, F. K. Butt, G. Shen, X. Zhang, S. X. Dou and J.-J. Zou, J. Mater. Chem. A, 2016, 4, 12940–12946 RSC.
- X. Han, J. Yang, C. Zhao, H. Huang, Z. Liu, P. M. Ajayan and J. Qiu, Adv. Mater. Interfaces, 2016, 8, 1500782 CrossRef.
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef CAS PubMed.
- J. Li, J. Li, X. Zhou, Z. Xia, W. Gao, Y. Ma and Y. Qu, ACS Appl. Mater. Interfaces, 2016, 8, 10826–10834 CrossRef CAS PubMed.
- M. Arif, G. Yasin, M. Shakeel, X. Fang, R. Gao, S. Ji and D. Yan, Chem. – Asian J., 2018, 13, 1045–1052 CrossRef CAS PubMed.
- J. Jiang, S. Lu, W.-K. Wang, G. Huang, B.-C. Huang, F. Zhang, Y.-J. Zhang and H.-Q. Yu, Nano Energy, 2018, 43, 300–309 CrossRef CAS.
- M. Shakeel, M. Arif, G. Yasin, B. Li, A. U. Khan, F. Khan and M. Baloch, Electrochim. Acta, 2018, 268, 163–172 CrossRef CAS.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488–2491 CrossRef CAS PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- C. Xiao, Y. Li, X. Lu and C. Zhao, Adv. Funct. Mater., 2016, 26, 3515–3523 CrossRef CAS.
- Y. Tang, X. Fang, X. Zhang, G. Fernandes, Y. Yan, D. Yan, X. Xiang and J. He, ACS Appl. Mater. Interfaces, 2017, 9, 36762–36771 CrossRef CAS PubMed.
- Z. Tao, T. Wang, X. Wang, J. Zheng and X. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35390–35397 CrossRef CAS PubMed.
- L. Yao, D. Wei, D. Yan and C. Hu, Chem. – Asian J., 2015, 10, 630–636 CrossRef CAS PubMed.
- Y. Li, Z. Y. Fu and B. L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., 2016, 55, 6290–6294 CrossRef CAS PubMed.
- J. Low, S. Cao, J. Yu and S. Wageh, Chem. Commun., 2014, 50, 10768–10777 RSC.
- Z. Haider and Y. S. Kang, ACS Appl. Mater. Interfaces, 2014, 6, 10342–10352 CrossRef CAS PubMed.
- M. Marelli, A. Naldoni, A. Minguzzi, M. Allieta, T. Virgili, G. Scavia, S. Recchia, R. Psaro and V. Dal Santo, ACS Appl. Mater. Interfaces, 2014, 6, 11997–12004 CrossRef CAS PubMed.
- Z. Wu, Z. Wang and F. Geng, ACS Appl. Mater. Interfaces, 2018, 10, 8585–8593 CrossRef CAS PubMed.
- M. Li, K. Chang, T. Wang, L. Liu, H. Zhang, P. Li and J. Ye, J. Mater. Chem. A, 2015, 3, 13731–13737 RSC.
- Y. Pi, Q. Shao, P. Wang, F. Lv, S. Guo, J. Guo and X. Huang, Angew. Chem., 2017, 56, 4502–4506 CrossRef CAS PubMed.
- L. Yao, N. Zhang, Y. Wang, Y. Ni, D. Yan and C. Hu, J. Power Sources, 2018, 374, 142–148 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- S. Wan, J. Qi, W. Zhang, W. Wang, S. Zhang, K. Liu, H. Zheng, J. Sun, S. Wang and R. Cao, Adv. Mater., 2017, 29, 1700286 CrossRef PubMed.
- M. Tahir, N. Mahmood, X. Zhang, T. Mahmood, F. K. Butt, I. Aslam, M. Tanveer, F. Idrees, S. Khalid, I. Shakir, Y. Yan, J. Zou, C. Cao and Y. Hou, Nano Res., 2015, 8, 3725–3736 CrossRef CAS.
- D. H. Youn, Y. B. Park, J. Y. Kim, G. Magesh, Y. J. Jang and J. S. Lee, J. Power Sources, 2015, 294, 437–443 CrossRef CAS.
- Z. Liu, C. Yu, X. Han, J. Yang, C. Zhao, H. Huang and J. Qiu, ChemElectroChem, 2016, 3, 850 CrossRef.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- P. F. Liu, S. Yang, B. Zhang and H. G. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 34474–34481 CrossRef CAS PubMed.
- J. Liu, J. Wang, B. Zhang, Y. Ruan, L. Lv, X. Ji, K. Xu, L. Miao and J. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 15364–15372 CrossRef CAS PubMed.
- Z. Wang, S. Zeng, W. Liu, X. Wang, Q. Li, Z. Zhao and F. Geng, ACS Appl. Mater. Interfaces, 2017, 9, 1488–1495 CrossRef CAS PubMed.
- W. Ye, X. Fang, X. Chen and D. Yan, Nanoscale, 2018, 10, 19484–19491 RSC.
- S. Bai, L. Wang, X. Chen, J. Du and Y. Xiong, Nano Res., 2015, 8, 175–183 CrossRef CAS.
- S. Bai, L. Yang, C. Wang, Y. Lin, J. Lu, J. Jiang and Y. Xiong, Angew. Chem., 2015, 54, 14810–14814 CrossRef CAS PubMed.
- Y. Zhao, X. Zhang, X. Jia, G. I. N. Waterhouse, R. Shi, X. Zhang, F. Zhan, Y. Tao, L.-Z. Wu, C.-H. Tung, D. O’Hare and T. Zhang, Adv. Energy Mater., 2018, 8, 1703585 CrossRef.
- Z. Chen, Y. Ha, Y. Liu, H. Wang, H. Yang, H. Xu, Y. Li and R. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 7134–7144 CrossRef CAS PubMed.
- L. Ge, Ch. Han, X. Xiao and L. Guo, Appl. Catal., B, 2013, 142-143, 414–422 CrossRef CAS.
- L. Ye, D. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 5280–5289 CrossRef CAS PubMed.
- B. Chai, T. Peng, J. Mao, K. Li and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 16745–16752 RSC.
- F. He, G. Chen, Y. Yu, S. Hao, Y. Zhou and Y. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 7171–7179 CrossRef CAS PubMed.
- H. Wang, X. Li and J. Yang, ChemPhysChem, 2016, 17, 2100–2104 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- H. Li, F. Zhao, J. Zhang, L. Luo, X. Xiao, Y. Huang, H. Ji and Y. Tong, Mater. Chem. Front., 2017, 1, 338–342 RSC.
- Y. Jia, J. Chen and X. Yao, Mater. Chem. Front., 2018, 2, 1250–1268 RSC.
- S. Zhang, J. Li, M. Zeng, G. Zhao, J. Xu, W. Hu and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12735–12743 CrossRef CAS PubMed.
- X. Zhou, B. Jin, R. Chen, F. Peng and Y. Fang, Mater. Res. Bull., 2013, 48, 1447–1452 CrossRef CAS.
- L. Ge, C. Han and J. Liu, Appl. Catal., B, 2011, 108, 100–107 CrossRef.
- Y. Yao, Y. Cai, F. Lu, J. Qin, F. Wei, C. Xu and S. Wang, Ind. Eng. Chem. Res., 2014, 53, 17294–17302 CrossRef CAS.
- M. Reli, P. Huo, M. Sihor, N. Ambrozova, I. Troppova, L. Matejova, J. Lang, L. Svoboda, P. Kustrowski, M. Ritz, P. Praus and K. Koci, J. Phys. Chem. A, 2016, 120, 8564–8573 CrossRef CAS PubMed.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. N. Waterhouse, Y. Zhao, C. Zhou, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- S. Martha, S. Mansingh, K. M. Parida and A. Thirumurugan, Mater. Chem. Front., 2017, 1, 1641–1653 RSC.
- S. Nayak, L. Mohapatra and K. Parida, J. Mater. Chem. A, 2015, 3, 18622–18635 RSC.
- W.-K. Jo and N. C. S. Selvam, J. Hazard. Mater., 2015, 299, 462–470 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- Z. Pan, Y. Zheng, F. Guo, P. Niu and X. Wang, ChemSusChem, 2017, 10, 87–90 CrossRef CAS PubMed.
- K. Liu, H. Zhong, F. Meng, X. Zhang, J. Yan and Q. Jiang, Mater. Chem. Front., 2017, 1, 2155–2173 RSC.
- M. Shakeel, M. Arif, G. Yasin, B. Li and H. D. Khan, Appl. Catal., B, 2019, 242, 485–498 CrossRef CAS.
- T. Bhowmik, M. K. Kundu and S. Barman, ACS Appl. Energy Mater., 2018, 1, 1200–1209 CrossRef CAS.
- L. Zhang, M. Ou, H. Yao, Z. Li, D. Qu, F. Liu, J. Wang, J. Wang and Z. Li, Electrochim. Acta, 2015, 186, 292–301 CrossRef CAS.
- Y. Hou, Z. Wen, S. Cui, X. Feng and J. Chen, Nano Lett., 2016, 16, 2268–2277 CrossRef CAS PubMed.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406–23433 RSC.
- C. G. Silva, Y. Bouizi, V. Fornes and H. Garcia, J. Am. Chem. Soc., 2009, 131, 13833–13839 CrossRef PubMed.
- Y. Lee, J. H. Choi, H. J. Jeon, K. M. Choi, J. W. Lee and J. K. Kang, Energy Environ. Sci., 2011, 4, 914–920 RSC.
- S. J. Kim, Y. Lee, D. K. Lee, J. W. Lee and J. K. Kang, J. Mater. Chem. A, 2014, 2, 4136–4139 RSC.
- Z. P. Xu and H. C. Zeng, Chem. Mater., 2001, 13, 4564 CrossRef CAS.
- P. Kustrowski, D. Sulkowska, L. Chmielarz, A. RafalskaLasocha, B. Dudek and R. Dziembaj, Microporous Mesoporous Mater., 2005, 78, 11 CrossRef CAS.
- R. Nakamura, A. Okamoto, H. Osawa, H. Irie and K. Hashimoto, J. Am. Chem. Soc., 2007, 129, 9596–9597 CrossRef CAS PubMed.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2014, 26, 2925–2930 CrossRef CAS PubMed.
- P. Chen, T. Y. Xiao, Y. H. Qian, S. S. Li and S. H. Yu, Adv. Mater., 2013, 25, 3192–3196 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00677f |
| This journal is © the Partner Organisations 2019 |