
Polyacryloyl hydrazide incorporation into ionic hydrogels improves toughness, elasticity, self-healability, adhesive & strain sensing properties†
Subhankar
Mandal‡
 a,
Niharika
Pandey‡
a,
Somendra
Singh
b,
Amit
Ranjan
b and
Umaprasana
Ojha
*a
a,
Niharika
Pandey‡
a,
Somendra
Singh
b,
Amit
Ranjan
b and
Umaprasana
Ojha
*a
aDepartment of Chemistry, Rajiv Gandhi Institute of Petroleum Technology, Jais, UP-229304, India. E-mail: uojha@rgipt.ac.in
bDepartment of Chemical Engineering, Rajiv Gandhi Institute of Petroleum Technology, Jais, UP-229304, India
First published on 25th February 2019
Abstract
Development of hydrogels possessing toughness, extensibility, resilience, and self-healability together is challenging. Herein, we demonstrate that incorporation of polyacryloyl hydrazide (PAHz) substantially improves the mechanical properties and incorporates self-healing, adhesive and strain sensing properties in ionic hydrogels. For example, the compressive strength (80 MPa), tensile strength (0.7 MPa) and fracture energy (∼902 J m−2) values of polyacrylamido-2-methylpropanesulfonic acid (PAMPS)-based hydrogels prepared by adding ∼6.5 wt% of PAHz increased by 73, 18 and 451 times, respectively, compared to that of the control. The presence of PAHz in the PAMPS hydrogel matrix provided additional physical crosslinking in the form of SO3−⋯H3N+–ionic interaction and created network reinforced zones that dissipated energy under stress and improved the toughness of the hydrogel system. At low water content (≤10 wt%), the compressive strength further increased to an unprecedented value of 0.4 GPa while maintaining adequate compressibility (92%). The PAHz incorporated-hydrogels exhibit an impressive adhesive strength value of up to 2.8 MPa on glass substrates, quick autonomous self-healing ability and strain sensing properties with a gauge factor value of up to 0.7. This general one-pot strategy may be extended to other ionic hydrogel systems for the improvement of mechanical properties.
Introduction
Synthetic hydrogels have shown promise in mimicking the mechanical abilities of natural tissues such as cartilage, tendons and ligaments.1,2 Specifically, several recently developed hydrogel systems possess low frictional coefficient, high compressive strength, extensibility, fracture strength, and wear resistance abilities. These hydrogels have also displayed promising self-healing,3 3D printing,4 water treatment,5 photo-voltaic,6 adhesive,7,8 controlled release,9,10 carbon capture,11 energy storage,12,13 biomedical,14 strain sensing,15 super adsorption16 and supercapacitor17 properties.18 Owing to the above, a lot of research effort is currently devoted to developing hydrogels with superior compressive, tensile and elastic properties. Among the several strategies used, the double-networking approach,19,20 use of nanoparticles as reinforcing agents,21 use of bulky groups,22 soaking-induced entanglement,23 use of low-melting-point alloy rigid scaffolds,24 orthogonal dual click approaches,25 topological slide rings,26 use of chemically crosslinked polymeric nanoparticles as crosslinking agents27 and the solvent-assisted conformational control strategy28 have been successful to develop hydrogels with improved mechanical properties.29,30 However, the synthetic procedures associated with some of these strategies require careful attention for large-scale material development, are multi-step in nature and require post-gelation processing to achieve the desired mechanical properties. Therefore, one-pot robust synthetic procedures for large scale synthesis of tough hydrogels possessing impressive mechanical properties are desirable.Polyacryloyl hydrazide (PAHz) is a water soluble polymer and possesses a weakly basic CONHNH2 functionality in each repeating unit. These CONHNH2 groups are capable of forming ionic linkages with various strongly acidic functionalities. Therefore, the presence of PAHz in ionic hydrogels possessing complementary anionic functionalities such as –SO3H, –COOH etc. was anticipated to provide additional physical crosslinking via ionic interactions and alter the mechanical properties. In fact, physical interactions such as ionic31 and hydrogen bonding linkages are known to supplement the mechanical properties by dissipating energy under load through reversible rupture and re-formation.32–34 Combination of ionic and chemical crosslinking has already been utilized as a design strategy to improve the mechanical properties of synthetic hydrogels.35,36 However, most of the literature efforts on ionically crosslinked hydrogels are limited to the use of metal ions (Ca2+, Fe3+etc.) or strongly cationic polyelectrolytes (unsubstituted and substituted amines, pKa ≈ 9–11).37,38 In this report, we utilize a weak cationic polyelectrolyte (PAHz) based on the CONHNH2 group (pKa ≈ 3–4) and study the effect of ionic complexation between CONHNH2 and a strongly anionic functionality (–SO3H) on the mechanical properties of the resulting dual crosslinked hydrogels.39 To the best of our knowledge, incorporation of PAHz to improve the mechanical properties of ionic hydrogels is rarely explored in the literature.
To evaluate the viability of the above strategy, polyacrylamido-2-methyl-1-propanesulfonic acid (PAMPS) possessing a complementary functionality (–SO3H) to –CONHNH2 was chosen as the ionic hydrogel system and the effect of PAHz incorporation on the mechanical properties of PAMPS was explored using a one-pot synthetic procedure. PAMPS is prolifically used in the literature to develop hydrogel systems for biomedical,40 membrane41,42 and other applications.43 The possible presence of physical interactions such as ionic linkages and hydrogen bonds in the resulting hydrogel systems raised the possibility of self-healing from damage. Moreover, the –SO3H and –CONH– functionalities present in the hydrogels were presumed to facilitate the interaction with various substrates and strong interaction inside the system through both chemical and physical crosslinks rendering these hydrogel systems suitable for use as adhesives. Therefore, the adhesive and strain sensing properties of these PAHz modified ionic hydrogel systems were explored and the adhesive efficiency was compared with several recently reported hydrogel adhesive systems.
Instrumentation
The compression studies of the hydrogel samples were recorded with cylindrical specimens (diameter 7 mm and length 15 mm). Hydrogel samples were obtained from the preparatory syringes and directly used for measurements. The water content of the hydrogels after removing them from the syringes was measured to be ∼70 wt%. The data were recorded at a machine speed of 2 mm min−1 using H25KS/H5KL, Tinius Olsen machines. The tensile tests were conducted using rectangular strips (30.0 × 9.5 × 2.0 mm3) of the hydrogels at a strain rate of 50 mm min−1. The samples for this were prepared by pouring the precursor solutions into molds made up of two glass slides separated by a 2 mm thick spacer. The tests were carried out within 5 h of removing the samples from the mold. The Young's modulus (E) values were calculated form the Hookean slope. The FT-IR spectra of the hydrogel samples were recorded using a PerkinElmer Spectrum Two FT-IR spectrometer. The equipment consisted of a PIKE MIRacle single reflection horizontal ATR accessory fitted with a ZnSe ATR crystal. Field-emission scanning electron microscopy (FESEM) images of the hydrogel samples were recorded using a Carl Zeiss-Sigma field-emission microscope (acceleration voltage ≈ 3 kV). The hydrogel samples were swelled in distilled water and freeze-dried under vacuum. The dried samples were cut into thin slices, gold coated and images were recorded. An advanced highly sensitive confocal fluorescence microscope LSM780NLO, Carl Zeiss GmbH system was used for imaging of the fluorescent dye tagged hydrogel samples. Images were processed using the ZEN 2012 software associated with the Carl Zeiss system. The average size distribution of the aggregates in the PAHz and AMPS aqueous solutions was measured by the dynamic light scattering method (DLS) using a Malvern's Zetasizer Nano-ZS equipped with a green laser of 523 nm. The DLS data presented is an average of three measurements. The Zetasizer software 6.20 (Malvern Instruments) was used to process the recorded data. A Magnus upright light microscope was used to monitor the self-healing efficiency of the hydrogels. For microscopy image acquisition and image processing the Magnus pro 3.7 software was used. The molecular weight of the polymer was measured by size exclusion chromatographic (SEC) analysis using a Waters system equipped with a 515 HPLC pump, an auto sampler, a Styragel column and a refractive index detector. THF was used as the eluent for the SEC analysis.Experimental
Materials
Poly(ethyleneglycol)diacrylate, Mn = 250 (PEGDA, Sigma Aldrich, 99%), AMPS (Sigma Aldrich, 99%), α-ketoglutaric acid (Sigma Aldrich, 99%), dansyl chloride (DC, Alfa Aesar, 97%), N,N′-methylene bisacrylamide (MBA, Sisco Research Lab., 99%), and hydrazine hydrate (NH2NH2·H2O, Fisher Scientific, 99%) were used as received. PAHz (Mn = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, PDI = 1.9) was synthesized following a reported procedure.44
000 g mol−1, PDI = 1.9) was synthesized following a reported procedure.44
Synthesis of PAHz incorporated hydrogels
To synthesize a typical PAHz incorporated hydrogel, 30 wt% aqueous solution of AMPS (12.0 mL, 17.0 mmol) was taken in a 30 mL glass vial and PEGDA (0.85 mmol) and 30 wt% PAHz (0.83 mL, 2.8 mmol) aqueous solution were added to it. This mixture was vortexed for 5 min and purged with N2 gas for 15 min. To this, α-ketoglutaric acid (0.17 mmol) was added with stirring. This solution was syringed out in 1 or 5 mL disposable syringes and kept under sun light or UV light (365 nm, light intensity ≈ 3.5 mW cm−2) for 6 h. The sealed samples were then stored for 4 days at 25 °C before analysis. Similarly, the PAMS-PAH-MBA (AMPS + PAHz + MBA) and PAMS-PAH-PEG (AMPS + PAHz + PEGDA) hydrogels were synthesized by varying the crosslinker type and amount in solution (ESI,† Table S1).Dye labelling of the hydrogel sample
Thin slices of PAHz incorporated hydrogel samples were dipped into a 2 wt% aqueous solution of DC for 10 h at 25 °C. The dye modified samples were removed from the DC solution and repeatedly washed with distilled water to remove the physically adsorbed dye molecules on the hydrogel surface. The samples were then dried on a smooth microscopy slide under ambient conditions. The confocal images of the dried film were recorded and analyzed.Calculation of the anisotropic swelling parameter
A dry hydrogel sample (0.6 × 0.4 × 0.1 cm3) was completely swelled in distilled water for 48 h and the dimensions were measured. The anisotropic swelling factor was calculated by using the following equation,where, H and H0 are the heights and L and L0 are the lengths of the rectangular hydrogel sample after and before swelling, respectively, measured by a slide caliper under a microscope (Fig. S1, ESI†).
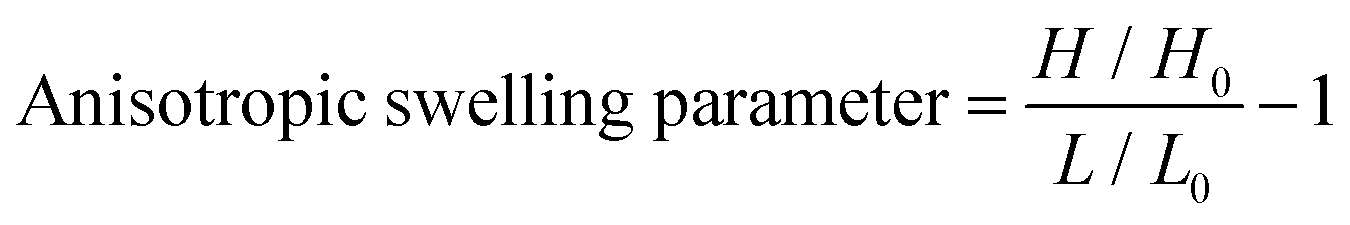
Hysteresis studies
Compressive loading–unloading curves of the hydrogel samples were recorded under controlled strain mode. Compressive strain from 0 to 75% was applied on the cylinder-shaped hydrogel samples containing 70 wt% water. The hysteresis energy (Eh, energy dissipated during a compressive loading–unloading cycle) was calculated using the area between loading and unloading curves of the hysteresis cycle using the Origin Pro 8.5 software.Fracture energy calculation
Fracture energy (G) of the hydrogels was determined by tearing test performed in a H5KL universal tensile machine (Tinius Olsen). A notch of 20 mm length was made in the rectangular hydrogel samples (width 5 mm, thickness 5 mm and length 30 mm) as shown in Fig. S2 (ESI†). The two legs of the sample were wrapped with cellulose-based adhesive tape to resist stretching during measurement and clamped up to the notch. The upper leg was pulled upward at a constant velocity of 5 mm min−1. The average tearing force “F” was recorded and “G” was calculated by using the equation; G = Favg/2w, where Favg and w are the average force and width of the hydrogel, respectively.Measurement of adhesion strength
Both the wet and dry lap-shear adhesive strength of the hydrogels were measured under tensile mode using a Tinius Olsen H5KL machine with 1000 N load cell and 10 mm min−1 load rate. For wet adhesive strength measurement, different hydrogel samples (containing 60 wt% water) were cut into thin sheets (1.0 × 0.5 × 0.1 cm3). A typical hydrogel sample was sandwiched between two substrates and pressed under a load of 500 g for 20 min before measurement. For dry adhesive strength measurement, the sandwiched substrates were incubated at 50 °C for 24 h under 500 g load. The adhered substrates were then used to record the adhesive strength values as per the schematics shown in Fig. S3 (ESI†).Strain sensing set-up
A typical hydrogel sample (50 wt% water content) possessing a dimension of 5.0 × 2.0 × 0.3 cm3 was clamped by metal binder clips at both sides and connected to a circuit comprising a red LED light indicator powered by a DC current source of 10 volt (Fig. S4, ESI†). The hydrogel was subjected to a number of stretching–releasing cycles and the changes in resistance (monitored by a digital multimeter connected to the circuit) along with the change in LED light intensity was recorded. To sense the motion of the human body, the hydrogel sample was fixed on a human index finger. Copper wires were attached to both ends of the hydrogel using Band-Aids. The wires were then connected to the circuit and resistance was monitored with different motions of the finger with time (Fig. S5, ESI†).Results and discussion
Various hydrogel compositions based on AMPS monomer and MBA/PEGDA crosslinker were prepared by adding PAHz in certain weight proportions (1.5–12.0 wt% of total solid content) (Scheme 1 and Table 1). The gelation was carried out via a one-pot strategy by adding the initiator (α-ketoglutaric acid) to the mixture of crosslinker, PAHz, and monomer in distilled water and exposing the resulting aqueous mixture to sun light or a UV source (365 nm). Sun light being a sustainable energy source was also used for the hydrogel synthesis and the properties were compared to that of the samples synthesized under UV light. The samples appeared cloudy during the crosslinking stage and the transparency improved gradually by storing the sealed hydrogels for 4 days under ambient conditions (Fig. S6, ESI†).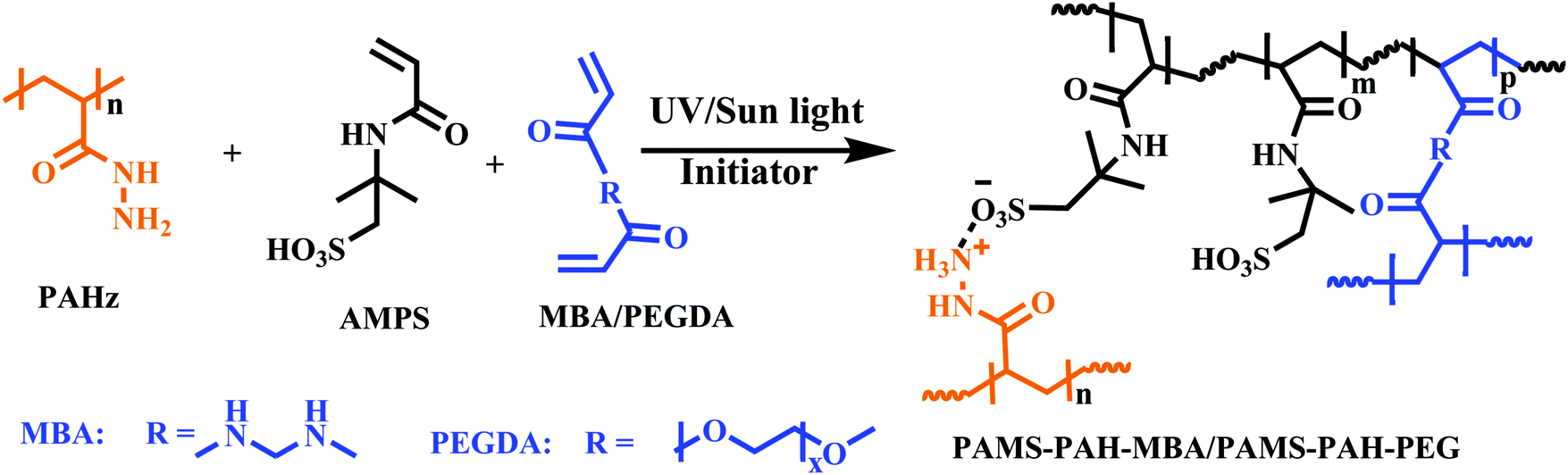 | ||
| Scheme 1 Synthesis of AMPS-based hydrogels using PAHz and PEGDA or MBA as the ionic and covalent crosslinkers, respectively. | ||
| Samplea | σ max* (MPa) | UTSb (MPa) | λ max (%) | E (MPa) | U max (MJ m−3) | G (J m−2) |
|---|---|---|---|---|---|---|
| a The as-synthesized samples possessed ∼70 wt% water and the water contents of the hydrogel samples were measured to be around 67 ± 3 wt%, while recording the mechanical properties. The mol% of the PAHz repeating unit was fixed at 14 mol% of total solid content for all samples except PAMS-MBA. PAMS-PAH-PEG′ and PAMS-PAH-MBA′ were prepared under UV light (365 nm) and PAMS-PAH-PEG and PAMS-PAH-MBA were prepared under sun light. b The ultimate tensile stress (UTS), E and ultimate elongation (λmax) values were determined from uniaxial tensile analysis. c The hydrogel possessed 1 mol% of MBA. d The hydrogel possessed 5 mol% of PEGDA with respect to AMPS, respectively. e Toughness (Umax) was determined from the σ versus strain curves. f Fracture energy (G) values were determined from the tensile tearing test. | ||||||
| PAMS-PAH-MBAc | 80.1 ± 1.5 | 0.7 ± 0.2 | 710 ± 12 | 0.4 ± 0.10 | 1.7 ± 0.3 | 902 ± 3 |
| PAMS-PAH-PEGd | 59.1 ± 2.5 | 0.9 ± 0.1 | 900 ± 23 | 1.1 ± 0.08 | 1.2 ± 0.4 | 1210 ± 6 |
| PAMS-MBAc | 1.1 ± 0.2 | 0.04 | 250 ± 25 | 0.06 ± 0.01 | 0.04 | 2 ± 1 |
| PAMS-PAH-PEG′d | 55.2 ± 3.6 | 1.1 ± 0.2 | 910 ± 15 | 1.0 ± 0.10 | 1.4 ± 0.1 | 1240 ± 5 |
| PAMS-PAH-MBA′c | 82.3 ± 2.1 | 0.8 ± 0.1 | 730 ± 18 | 0.4 ± 0.07 | 1.7 ± 0.8 | 980 ± 3 |
The domains in the resulting hydrogel matrix possessing PAHz were anticipated to be network reinforced zones owing to the additional physical crosslinking provided by the CONHNH3+⋯SO3− ionic linkages (Fig. 1). The pair of FTIR bands at 1232 and 1076 cm−1 accountable to SO3H of AMPS shifted to 1160 cm−1 in the PAHz-AMPS mixture supporting ionization of –SO3H to –SO3− (Fig. S7, ESI†). Similarly, the band at 3275 cm−1 for –NH2 in PAHz shifted to 3288 cm−1 in the hydrogel suggesting possible formation of CONHNH3+ ions.
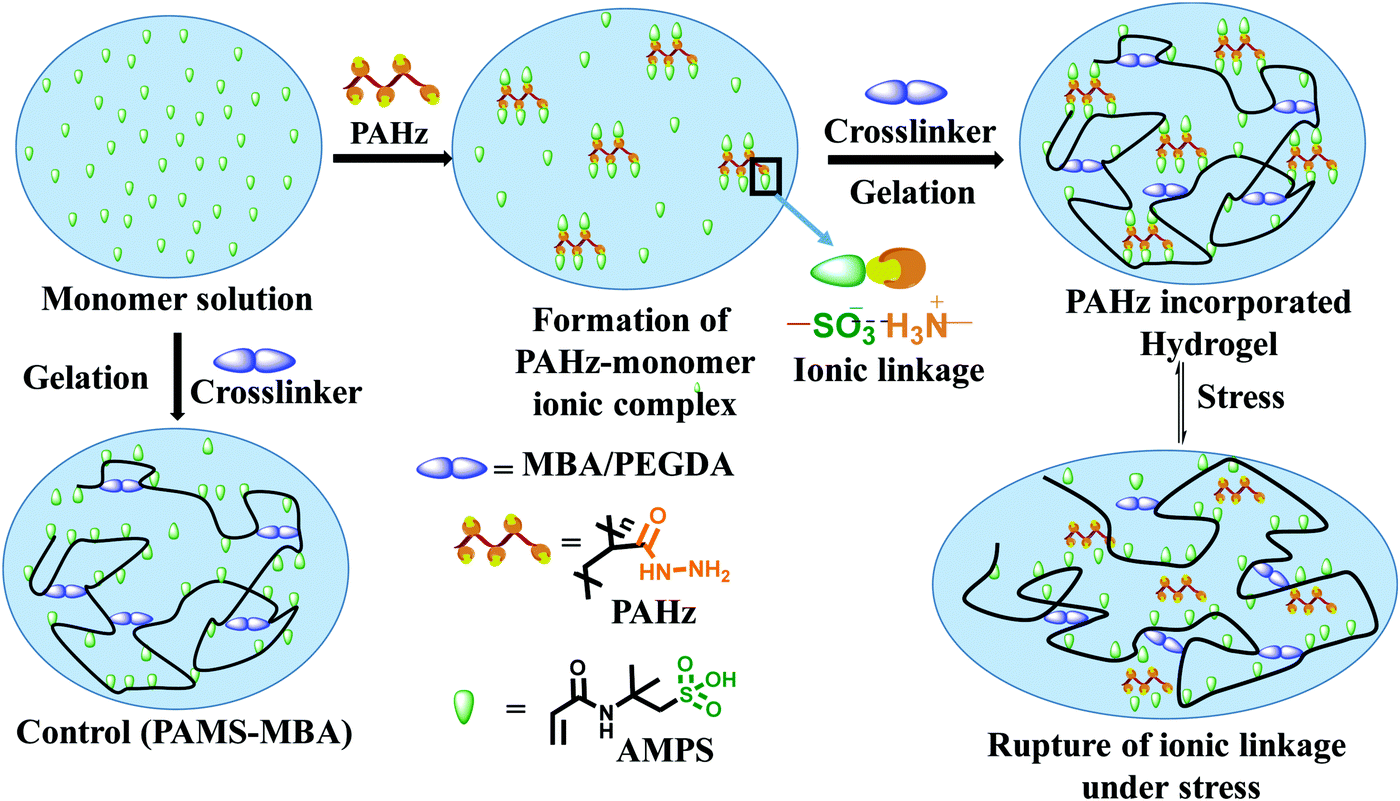 | ||
| Fig. 1 Schematics showing the synthesis of control and PAHz incorporated hydrogels and the mechanism of energy dissipation under stress. | ||
To visualize these possible PAHz mediated ionic complex zones in the hydrogel matrix, a dye labeling technique was used. DC was chosen as the dye for this purpose, since the dye is capable of reacting with the PAHz under ambient conditions and exhibiting fluorescent properties after structural modification (Scheme S1, ESI†).45 Fluorescence confocal microscopy images of a cross-sectional view of the DC labelled PAMS-PAH-MBA revealed fluorescent patches of irregular shapes with average sizes in the range of 0.6 to 1.2 μm dispersed throughout the matrix (Fig. 2). These micron size fluorescent patches in the hydrogel matrix were attributed to the ionic complex zones in the samples. Possibly, the extent of ionic complexation between the PAHz and AMPS monomers controlled the sizes of these ionic complex domains.
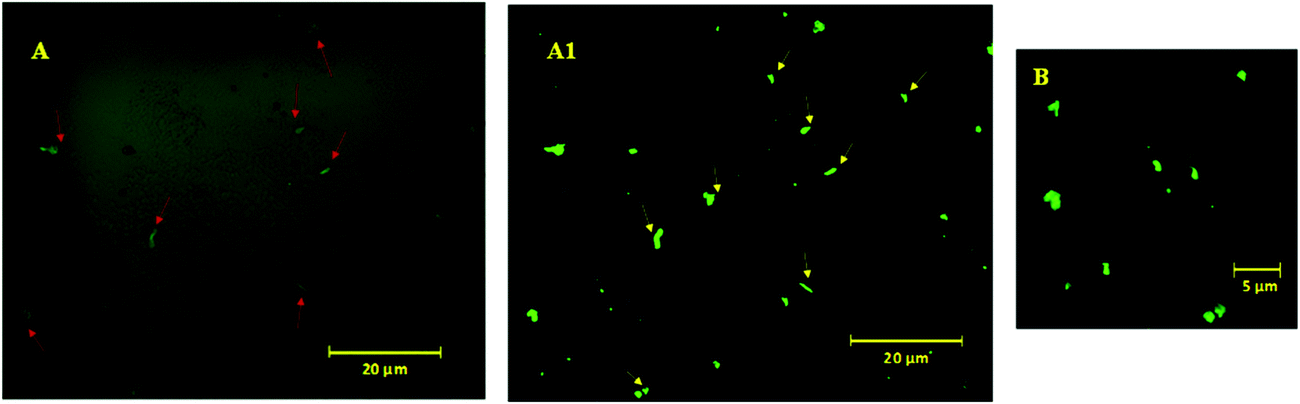 | ||
| Fig. 2 Confocal fluorescence microscopy images of DC-tagged cross-sectional view of PAMS-PAH-MBA hydrogel under different magnifications (A, A1 & B). A1 was obtained after background processing of (A) using the ZEN 2012 software. The red and yellow colored arrows in figures (A) and (A1) show the PAHz incorporated zones in the hydrogel matrix. | ||
To further support the domain sizes of these ionic crosslinked zones, DLS data of PAHz solution in the presence of different concentrations of AMPS were recorded. The DLS and FESEM data revealed that, PAHz (2 wt%) in aqueous solution formed aggregates of ∼20 nm size. In the presence of AMPS the average size gradually increased up to ∼70 nm and new peaks around 200 nm and ∼2 μm appeared (Fig. S8, ESI†). This supported the confocal data and the sizes of these ionic micro-domains in the hydrogels resulting from the ionic complexation of PAHz and AMPS. The swelling parameter (∼0.14) value calculated based on reported procedures revealed a certain degree of anisotropic swelling characteristics in rectangular PAMS-PAH-MBA samples (Fig. S1, ESI†).46 This anisotropic swelling of the samples supported the presence of strongly crosslinked zones in the hydrogel matrix.47 At the initial stages of swelling (within 10 min), the PAMS-PAH-MBA hydrogel displayed an uneven swelling pattern and the appearance of curly textures on the swelled surface, which became uniform with an increase in water content (Fig. S9, ESI†). The presence of strongly crosslinked domains in the light crosslinked matrix is known to induce non-axisymmetric swelling patterns in the samples.48
The amounts of PAHz and crosslinker in the hydrogel compositions were optimized based on their σmax values. The samples possessing 14 mol% (with respect to repeating unit) of PAHz and 1 mol% of MBA or 5 mol% of PEGDA exhibited maximum σmax value in all cases (Fig. 3A and B). The σmax values of PAMS-PAH-MBA (∼80.1 MPa) and PAMS-PAH-PEG (∼59.1 MPa) strongly improved compared to that of the PAMS-MBA (1.1 MPa). The σmax values of the PAMS-PAH-MBA′ (82.3 MPa) and PAMS-PAH-PEG′ (55.2 MPa) samples were comparable to those of the samples synthesized under sun light (Table 1). Therefore, these selective compositions were used for further studies (Table 1). The σmax value is superior compared to that of the several tough hydrogels reported in the literature.
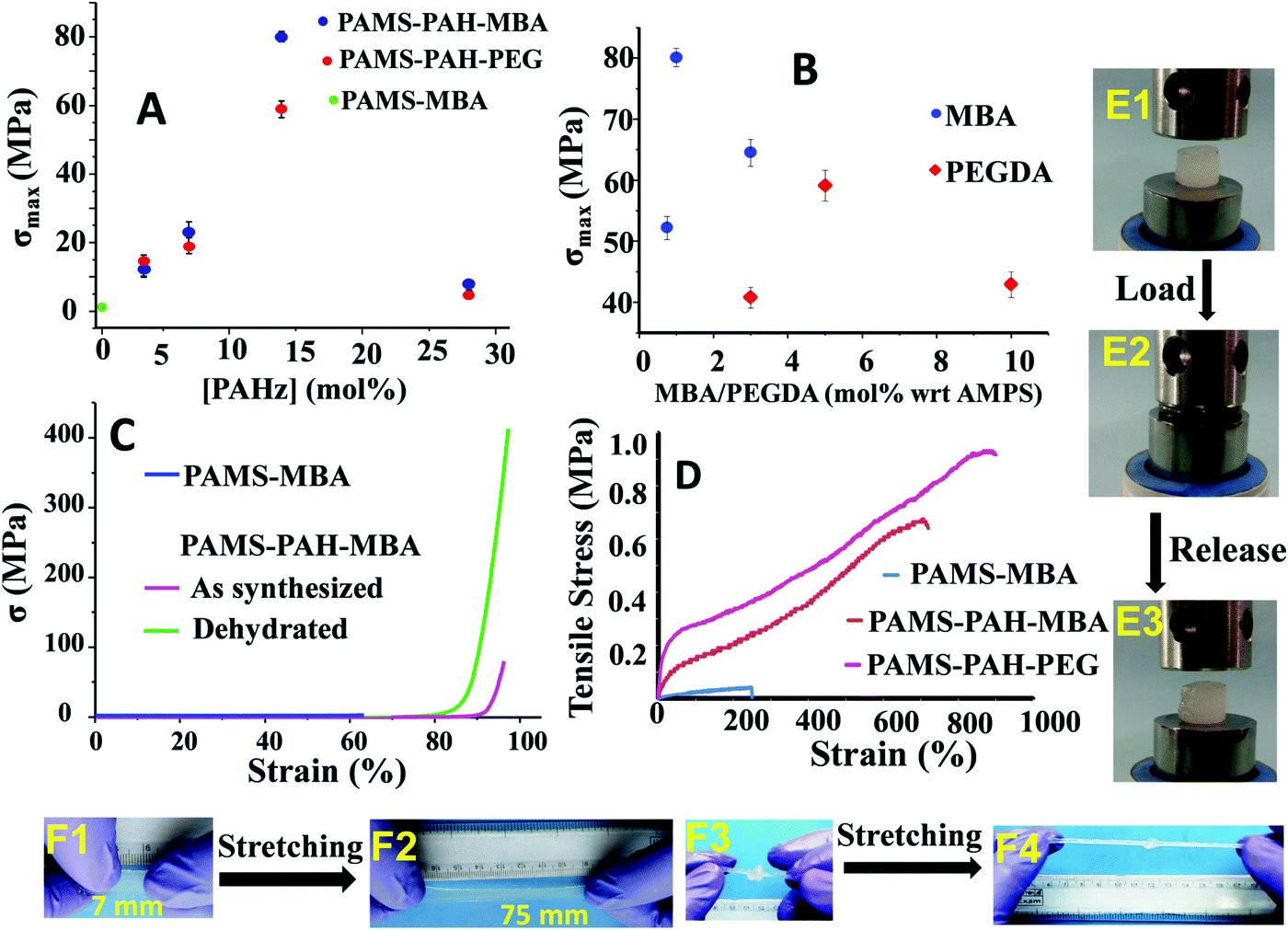 | ||
| Fig. 3 Effect of (A) PAHz amount and (B) crosslinker mol% with respect to AMPS on the σmax of the hydrogels, (C) the σ versus strain plots of as-synthesized (water content ≈70%) and dehydrated (water content ≈10%) PAMS-PAH-MBA, (D) tensile traces of the hydrogels, (E1) a cylindrical PAMS-PAH-MBA sample possessing 70 wt% water, (E2) E1 compressed to 90% strain, (E3) the sample after release of the load, (F1) a thin PAMS-PAH-MBA strip, (F2) F1 stretched to ∼900% of its original size, (F3) a knotted PAMS-PAH-MBA cylindrical sample, and (F4) the knotted sample stretched to 500%. | ||
The Umax value of the PAMS-PAH-MBA (1.7 MJ m−3) was 40 times higher compared to that of the control (0.04 MJ m−3) (Table 1). A PAMS-PAH-MBA sample (water content ≈ 70 wt%) compressed to ∼90% of its original height quantitatively recovered on release of the stress, whereas PAMS-MBA broke into multiple pieces (Fig. 3E & Fig. S10, ESI†). Possibly, the ionic linkages present in the dual crosslinked hydrogel dissipated energy through reversible rupture under stress and strongly improved the compressive properties as envisaged (Fig. 1). High mechanical dissipation in the systems is important to enhance the toughness of the samples.49
To further support our claim, the hysteresis data of PAMS-PAH-MBA were compared to that of the control. These PAHz incorporated hydrogels exhibited remarkable resilience and anti-fatigue properties (Fig. 4A). PAMS-PAH-MBA recovered up to 99% of the original shape after five consecutive cycles and the recovery time (∼2 min) after each cycle was relatively faster compared to that of the already reported tough hydrogels.50,51 The loading and unloading curves almost superimposed upon each other and no residual strain was noticed at the end of each unloading cycle. The dissipated energy of PAMS-PAH-MBA was ∼138 times higher compared to that of the PAMS-MBA supporting our design strategy that the ionic linkage-embedded micro-domains present in the PAHz incorporated hydrogels are responsible for the energy dissipation (Fig. 4B & Fig. S11, ESI†).
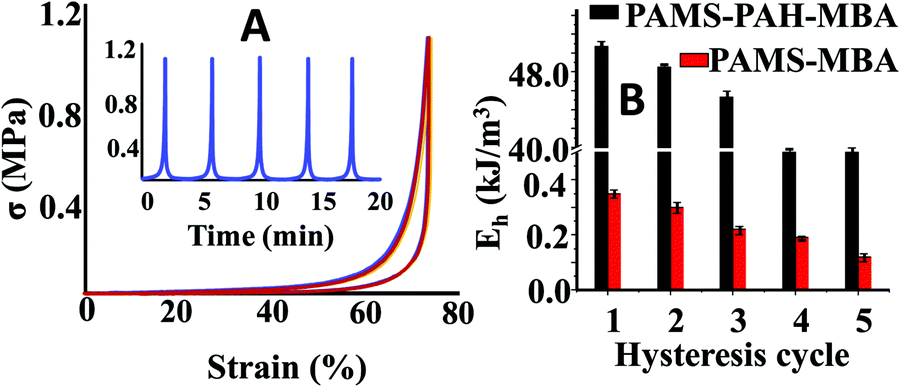 | ||
| Fig. 4 (A) Strain controlled hysteresis data of PAMS-PAH-MBA. (B) Comparison of energy of dissipation between PAMS-PAH-MBA and PAMS-MBA for different hysteresis cycles. | ||
PAMS-MBA simply broke on a single impact with a hammer, whereas the PAMS-PAH-MBA survived multiple impacts without any noticeable deformity (Movie S1, ESI†). Importantly, the sample instantly recovered the original shape each time after the impact. The sample resisted repeated attempts of cutting using a blunt kitchen knife and remained intact after being run over multiple times by a car (car weight ≈ 1.3 ton) (Movies S2 and S3, ESI†). These demonstrations revealed that the PAHz incorporated-hydrogels are resilient and suitable for high impact applications. The σmax value (∼59.1 MPa) of PAMS-PAH-PEG was somewhat lower than that of the PAMS-PAH-MBA (80.1 MPa) (Table 1). This may be attributed to the compact network structure of the latter resulting from the short chain crosslinker (MBA) (Fig. S12, ESI†). Interestingly, the dehydrated (water content ≤10 wt%) PAMS-PAH-MBA displayed an unprecedented σmax value (∼0.4 GPa), while maintaining excellent compressibility (92%) (Fig. 3C). The sample recovered the original shape within 1 h of releasing the stress. This suggested that these dried crosslinked networks may be used as oleophobic rubber for printing and other load bearing applications in organic media, where natural rubber plasticizes on long-term contact with organic solvents and oils. A simple demonstration shows that a piece of commercial latex rubber gradually disintegrated over 10 days of immersion in THF, whereas PAMS-PAH-MBA's shape and elasticity remained least affected by the same exposure (Fig. S13, ESI†).
The molar ratio between SO3H:CONHNH2 (0.86:0.14) in samples (PAMS-PAH-MBA & PAMS-PAH-PEG) possessing optimum mechanical properties was far from the charge balance point. Strong swelling ability of the hydrogels supported the charge imbalance in these hydrogel systems. Under equilibrium water uptake conditions, the water content of both PAMS-PAH-MBA and PAMS-PAH-PEG strongly increased up to 98.4 and 96.9 wt%, respectively. The σmax values of the equilibrated PAMS-PAH-MBA and PAMS-PAH-PEG hydrogels decreased to 1.2 and 2.1 MPa, respectively. Importantly, the values were notably higher compared to that of the equilibrated PAMS-MBA control (water content ≈ 98.2 wt%, σmax ≈ 0.006 MPa). Intuitively, the modulus value of dehydrated PAMS-PAH-MBA (0.46 MPa) was substantially higher compared to that of the equilibrated sample (0.005 MPa).
These PAHz incorporated-hydrogels displayed λmax and UTS values up to ∼900% and 0.9 MPa, respectively (Fig. 3D). The E values of these hydrogels were recorded in the range of 0.4 to 1.1 MPa, which is comparable to that of the pig bone cartilage,52 and somewhat higher than that of the skin53 as reported in the literature (Table 1). The UTS values of PAMS-PAH-MBA and PAMS-PAH-PEG were ∼18–23 times higher than that of the control. PAMS-PAH-MBA strips and knotted samples were reversibly stretched up to ∼500% without fracture (Fig. 3F). The G values of the PAHz incorporated-hydrogels (902–1210 J m−2) were substantially higher compared to that of the PAMS-MBA (2 J m−2) (Table 1). The UTS value (1.1 MPa) of PAMS-PAH-PEG′ was somewhat higher compared to that of the PAMS-PAH-PEG (0.9 MPa) (Table 1 and Fig. S14, ESI†). However, the PAMS-PAH-MBA samples prepared using a low amount of MBA (0.1 mol%) displayed substantially low UTS (0.1 MPa) and σmax (11.2 ± 3.1 MPa) values and the samples possessing 0.05 and 0.07 mol% of MBA displayed fluid-like behavior (Fig. S15, ESI†).
The mechanical properties of the PAHz-based PAMPS hydrogels were compared to those of the several recently reported tough hydrogel systems based on a dual (ionic and covalent) crosslinking approach (Table 2). The σmax (∼80 MPa), UTS (∼0.9 MPa) and E (∼1.1 MPa) values of the current hydrogels are comparable and in some cases superior to those of the existing systems (σmax ≈ 15 to 62 MPa, UTS ≈ 0.2 to 1.4 MPa, and E ≈ 0.009–1.0 MPa), whereas the G (≈1210 J m−2) and λmax (900%) values are inferior to some of the tough hydrogels reported earlier (λmax up to 2980%, G ≤ 8700 J m−2).
| Compositiona | σ max (MPa) | UTS (MPa) | λ max (%) | E (MPa) | G (J m−2) | Ref. |
|---|---|---|---|---|---|---|
| a The mechanical properties were measured for water contents in the range of 60–90 wt%. | ||||||
| Alginate (Ca2+)-PAAm | — | 0.16 | 2300 | 0.029 | 8700 | 32 |
| GO + PAA-Fe3+ | — | 0.77 | 2980 | — | — | 54 |
| PAA/SMA-Si NPs | — | 0.25 | 2823 | 0.009 | — | 55 |
| Silicate/PAMPS/PAAm IPN | 61.9 | 1.4 | 112 | 1.0 | — | 56 |
| κ-Carrageenan/PAAm | 15.4 | 0.56 | 2100 | 0.125 | 6150 | 57 |
| Agar/PAAc/Fe3+ | — | 0.32 | 1130 | 0.11 | 1015 | 58 |
| PAMS-PAH-PEG | 59.1 | 0.9 | 900 | 1.1 | 1210 | This work |
| PAMS-PAH-MBA | 80.1 | 0.7 | 710 | 0.4 | 902 | This work |
These hydrogels exhibited autonomous self-healing ability. A cylindrical PAMS-PAH-PEG specimen was cut into two pieces using a sharp blade and simply joined together with mild force and kept in a closed container at 25 °C for possible autonomous self-healing. The self-healing was efficient and the healed sample sustained stretching up to 500% after 5 min of healing period (Fig. 5B and Movie S4, ESI†). The healing efficiency was quantified based on the tensile data recorded with rectangular samples. The edges of the cut rectangular strips were touched with each other and left undisturbed for self-healing. A healing period of 2 h was sufficient to recover the UTS and λmax values up to ∼80 and 90%, respectively (Fig. 5A). The conductivity of PAMS-PAH-PEG hydrogels was measured using a four-probe set-up (Fig. S16, ESI†). The values of the as-synthesized (water content ≈ 70 wt%) and dehydrated (water content ≈10 wt%) samples were 1.7 and 0.6 S m−1, respectively. The conductivity of the samples may be attributed to the presence of sulfonic acid groups in the hydrogel matrix59 and the green LED connected through the PAMS-PAH-PEG strip was able to glow supporting the above (Fig. 5C1). The light extinguished on cutting the sample into two separate pieces. On joining the two pieces together the LED was lit again and the intensity of the light gradually improved with the self-healing period suggesting reinstatement of ionic conduction in the hydrogel matrix and self-healing of the hydrogel (Fig. 5C2 and C3). Joined PAMS-PAH-PEG cylindrical pieces were able to bear 230 g of load after 30 min of autonomous healing further supporting the quick self-healing ability of these hydrogel systems (Fig. 5D). The crack healing on the PAMS-PAH-PEG sample surface was monitored after regular time intervals under a microscope (Fig. 6A). Disappearance of the crack interface within 24 h revealed that the bonding is via self-healing. However, the self-healing of PAMS-PAH-MBA was slower compared to that of the PAMS-PAH-PEG hydrogel. The efficient self-healing in PAMS-PAH-PEG samples may be attributed to the presence of multiple physical interactions such as SO3−⋯H3N+ ionic linkage and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HN hydrogen bonds in the samples. Furthermore, the presence of PEG in the hydrogels resulted in an amphiphilic system consisting of immiscible PEG, PAMPS and PAHz segments. Interaction between chains of individual segments to phase-segregate also promoted chain diffusion and self-healing. Micelle formation in PEG-PAMPS block copolymers is already known in the literature.60 A plausible self-healing mechanism based on the reversible dissociation and re-formation of the above three interactions is schematically presented in Fig. 6B. Self-healable hydrogels based on hydrogen bonding,61,62 ionic interaction63,64 and segment association65 are already known in the literature.
O⋯HN hydrogen bonds in the samples. Furthermore, the presence of PEG in the hydrogels resulted in an amphiphilic system consisting of immiscible PEG, PAMPS and PAHz segments. Interaction between chains of individual segments to phase-segregate also promoted chain diffusion and self-healing. Micelle formation in PEG-PAMPS block copolymers is already known in the literature.60 A plausible self-healing mechanism based on the reversible dissociation and re-formation of the above three interactions is schematically presented in Fig. 6B. Self-healable hydrogels based on hydrogen bonding,61,62 ionic interaction63,64 and segment association65 are already known in the literature.
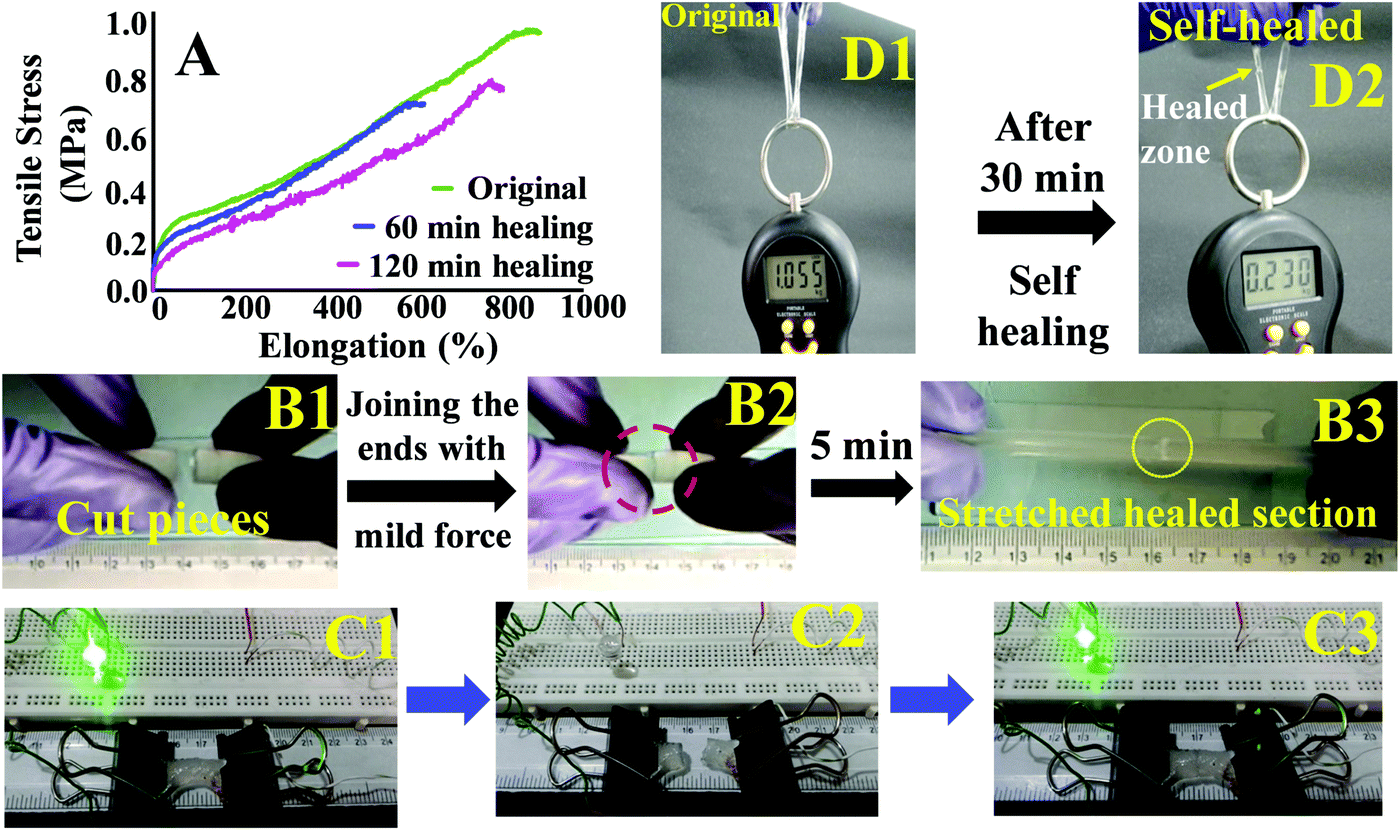 | ||
| Fig. 5 (A) Tensile traces of the original and self-healed PAMS-PAH-PEG after different time intervals, (B1) two cut pieces of PAMS-PAH-PEG, (B2) the cylindrical pieces joined together by mild force & (B3) the self-healed PAMS-PAH-PEG resisting stretching up to 500%. (C1) A circuit connected though a PAMS-PAH-PEG strip shows glowing of an LED, (C2) the circuit broke on cutting the hydrogel sample, and (C3) the LED glowed with similar intensity compared to that of C1 after 5 min self-healing of the hydrogel. (D1) A PAMS-PAH-PEG cylindrical strip bearing 1 kg load, and (D2) self-healed D1 bearing 230 g load after 30 min of healing time. | ||
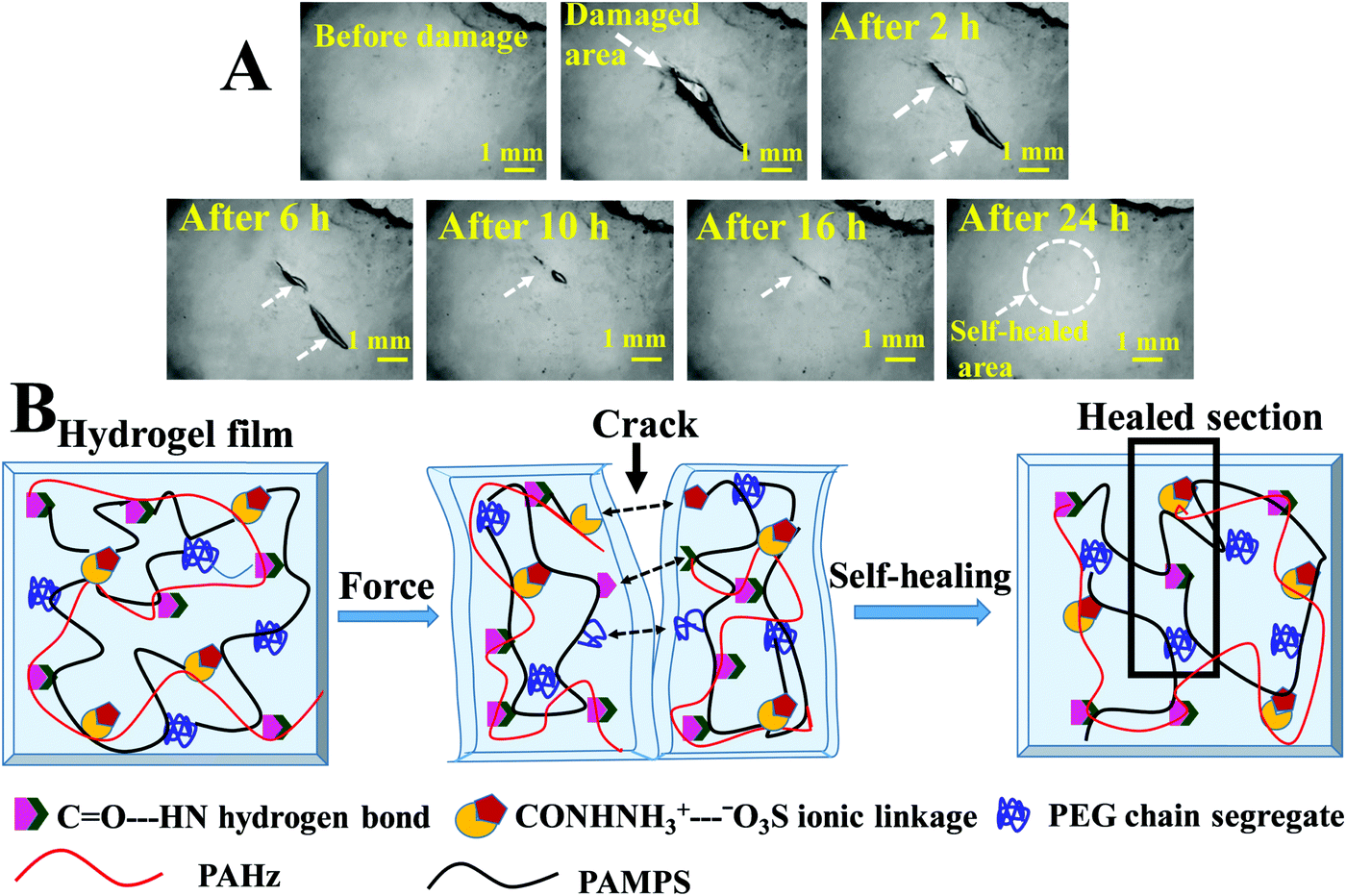 | ||
| Fig. 6 (A) Microscopy images showing the crack healing on the surface of PAMS-PAH-PEG hydrogel film possessing 60 wt% water (the samples were kept in a sealed glass container to resist evaporation of water during the self-healing process). (B) Schematics of self-healing in a PAMS-PAH-PEG hydrogel sample. | ||
The hydrogel samples exhibited impressive adhesive properties towards metal, skin, plastic, wooden and glass surfaces (Fig. 7A). A typical PAMS-PAH-PEG sample weighing 0.5 g was able to hold a 300 g glass object (Fig. 7A3). The adhesive strength values of the rectangular hydrogel samples were determined from the lap-shear tests. This procedure is regularly used in the literature to evaluate the adhesive efficiency of hydrogels.66,71 Moreover, the method allowed the measurement of both wet and dry adhesive strengths of the samples under similar conditions. A curing time of 20 min under 500 g load at 25 °C was sufficient for the PAMS-PAH-PEG to achieve maximum adhesive strength values on glass substrates (Fig. 7B). The adhesive strength values of PAMS-PAH-PEG for different surfaces were measured in the range of 40–110 kPa (Fig. 7D and Table S2, ESI†). The value was maximum for the glass substrate.
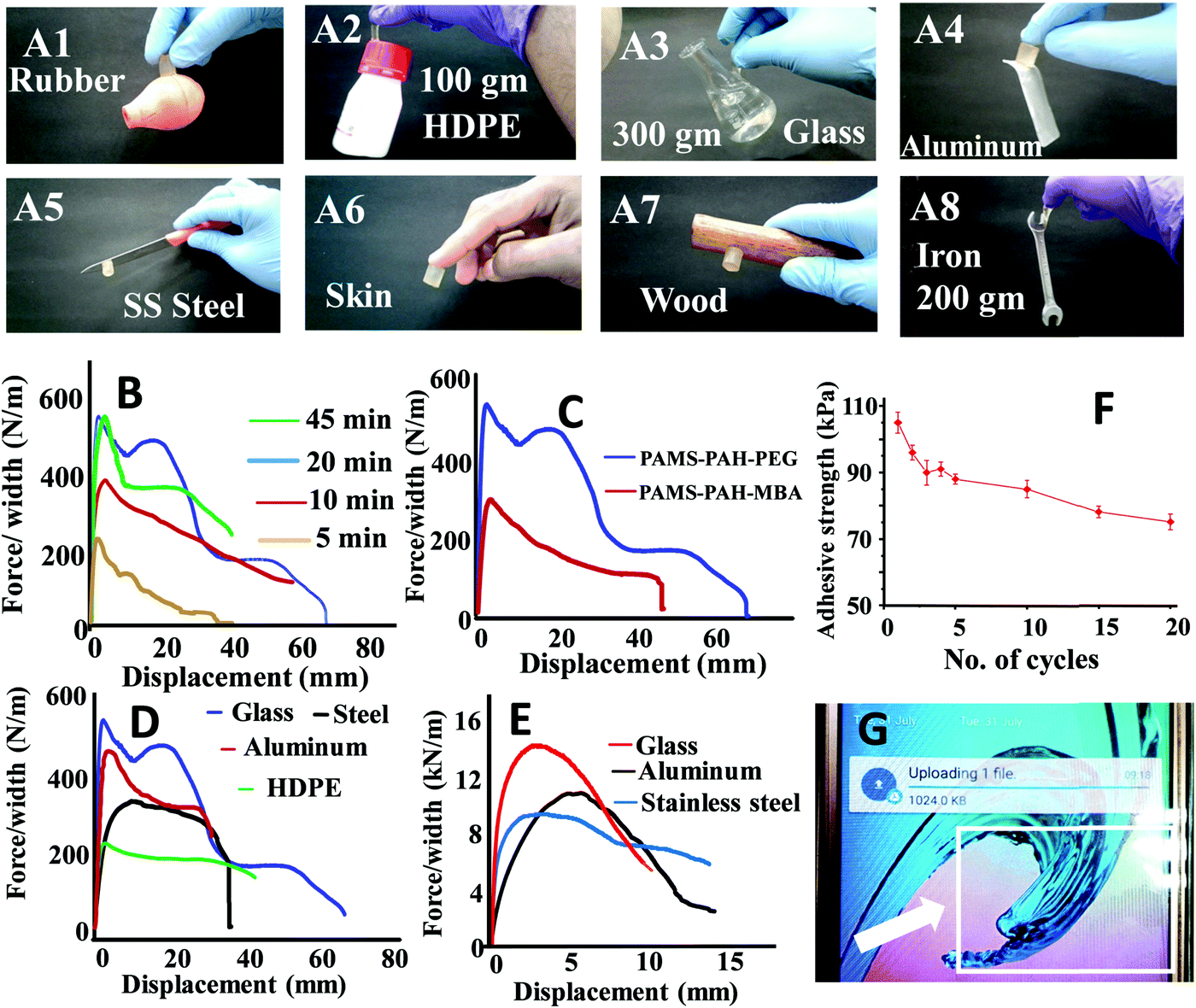 | ||
| Fig. 7 (A1–A8) Photographs showing the adhering efficiency of PAMS-PAH-PEG towards different substrates. The adhesive strength plots of (B) PAMS-PAH-PEG on a glass substrate after different time intervals, (C) different hydrogels on a glass substrate, (D) PAMS-PAH-PEG on different substrates and (E) dry PAMS-PAH-PEG on different substrates. (F) The adhesive strength of PAMS-PAH-PEG on glass after different cycles. (G) Photograph showing a screen guard pasted on a mobile phone screen using PAMS-PAH-PEG. | ||
A comparative analysis of the adhesive strength of these hydrogels with some of the recently reported hydrogel adhesives revealed that the adhesive efficiency of the current system on glass substrates is comparable to those of the several reported systems (Table 3).72,73 The high adhesive strength of PAMS-PAH-PEG towards glass may be assigned to the presence of various physical interactions such as electrostatic, van der Waals and hydrogen bonding interaction between the hydrogel and glass and cohesive interactions in the hydrogel matrix. The efficient recovery of the samples from high cyclic stress (∼0.2 MPa) supported the strongly cohesive nature of the PAMS-PAH-PEG (Fig. S17, ESI†). The reusability of the adhesive was monitored by repeatedly sandwiching a typical wet PAMS-PAH-PEG thin strip between two glass slides and measuring their adhesive strength until failure. The adhesive strength decreased by ∼28% only over 20 consecutive cycles (Fig. 7F). The minor decrease in the strength may be assigned to dust accumulation on the hydrogel surface due to repetitive handling. Nevertheless, the satisfactory wet adhesive strength value of 76 kPa after the 20th cycle suggested that PAMS-PAH-PEG may be utilized as a suitable reusable adhesive. The adhesive efficiency of PAMS-PAH-PEG (shear adhesive strength ≈ 0.8–2.8 MPa) increased by approximately ∼20 times for all substrates on evaporation of water (Fig. 7E). Possibly, on evaporation of water, the extent of physical interactions between the hydrogel and substrate increased, which strengthened the adhesion. The dry adhesive strength value of PAMS-PAH-PEG (2.8 MPa) on a glass surface is superior compared to that of the several recently reported systems (Table 3). The tack adhesive strengths of PAMS-PAH-MBA (0.02 MPa) and PAMS-PAH-PEG (0.04 MPa) were measured on a glass surface following a procedure similar to that reported in the literature (Fig. S18, ESI†).74 The superior value of shear adhesive strength (PAMS-PAH-PEG ≈ 0.11 MPa) compared to that of the tack test may be attributed to the strongly crosslinked nature and high modulus of the hydrogel samples.
The strain sensing ability of PAMS-PAH-PEG was accessed by observing the resistance change with increase in strain. At 0% strain, a resistance value of 3 kΩ was observed. The resistance increased by ∼700% on increasing the strain up to 900% (Fig. 8A). This may be attributed to the narrowing of the hydrogel cross section and increase in path length. The gauze factor value of PAMS-PAH-PEG is comparable to various hydrogel-based strain sensors reported in the recent literature.72,75 The resistance change was reversible and the initial resistance value was regained on releasing the strain. To understand the stability of the resistance change with respect to strain, the sample was reversibly extended to 100% strain for five continuous cycles and the resistance change was measured. Each time the resistance reached a value of 3.4 kΩ at 100% strain and the value decreased to 2.9 kΩ on releasing the strain (Fig. 8B). The resistance change was demonstrated by connecting a LED circuit through a PAMS-PAH-PEG strip. The intensity of light gradually decreased on increasing the strain of the PAMS-PAH-PEG strip and only a faint illumination was visible at 500% strain (Fig. 8C1–C3). The intensity of the LED was reinstated on gradually releasing the strain on the hydrogel (ESI,† Movie S5).
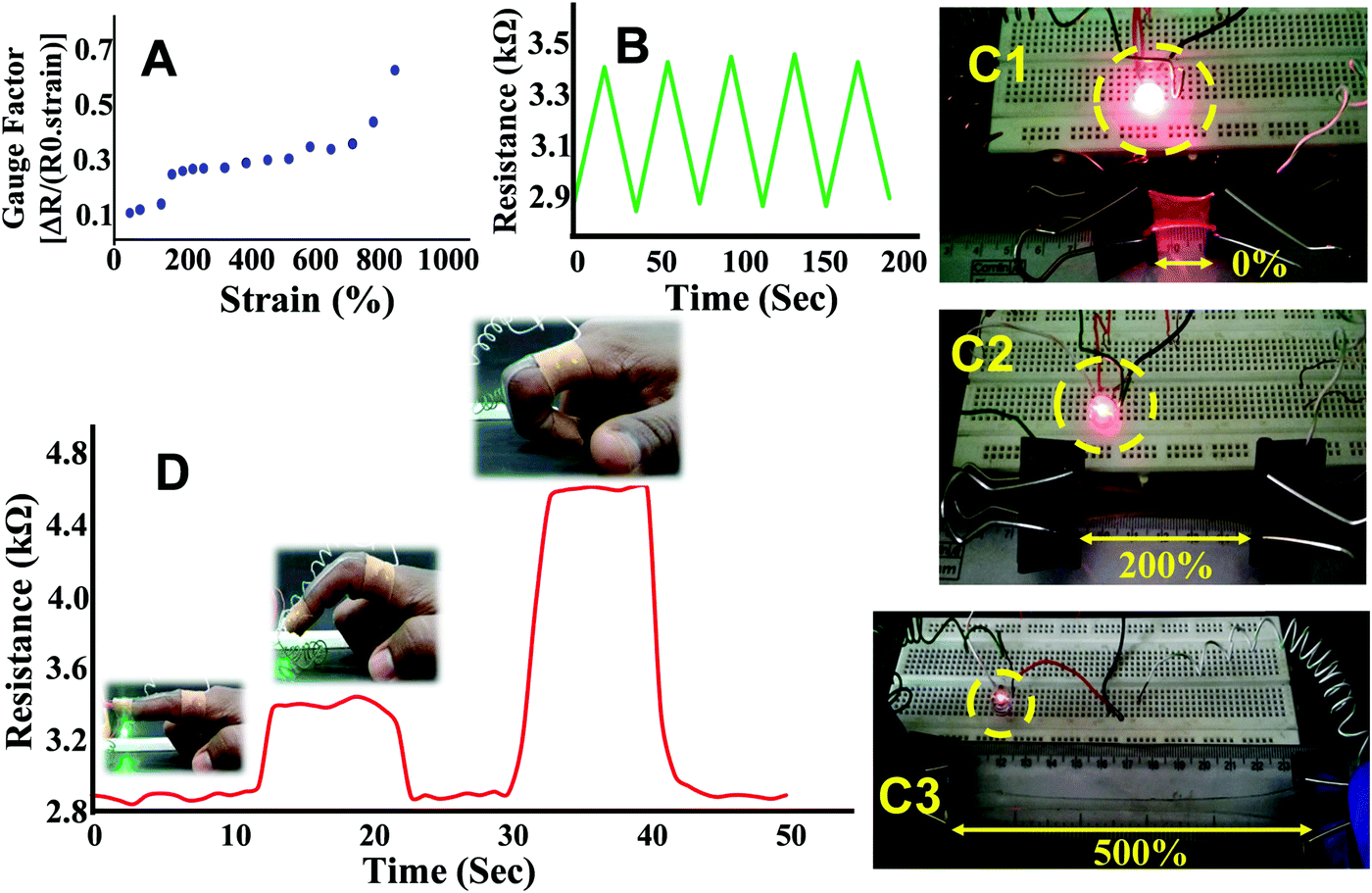 | ||
| Fig. 8 (A) The gauge factor versus strain (%) curve, (B) five consecutive cycles showing the variation in resistance with reversible stretching, (C1–C3) the change in illumination of a red LED with stretching of the hydrogel, and (D) a PAMS-PAH-PEG strip pasted on the finger and subsequently connected to a multimeter showing the change in resistance with the motion of the finger. | ||
To demonstrate real life applications of the hydrogel through sensing of the motions of the human body, a rectangular strip was attached to a human finger and the corresponding change in resistance with the movement of the finger was measured. The resistance gradually increased from 2.8 to 4.4 kΩ with bending of the finger suggesting that the hydrogel may potentially be used in a biomedical device to monitor various motions of the body (Fig. 8D). The baseline shift during the repeated measurements was minimal suggesting that the data collection was consistent. The sensing of movement of various parts of the body was further monitored by attaching PAMS-PAH-PEG strips to elbow and knee joint models (Fig. 9B & C). The resistance of the sample reversibly changed from 2.8 to 4.1 kΩ with gradual bending of the knee joint model (Fig. 9A and B1–B3).
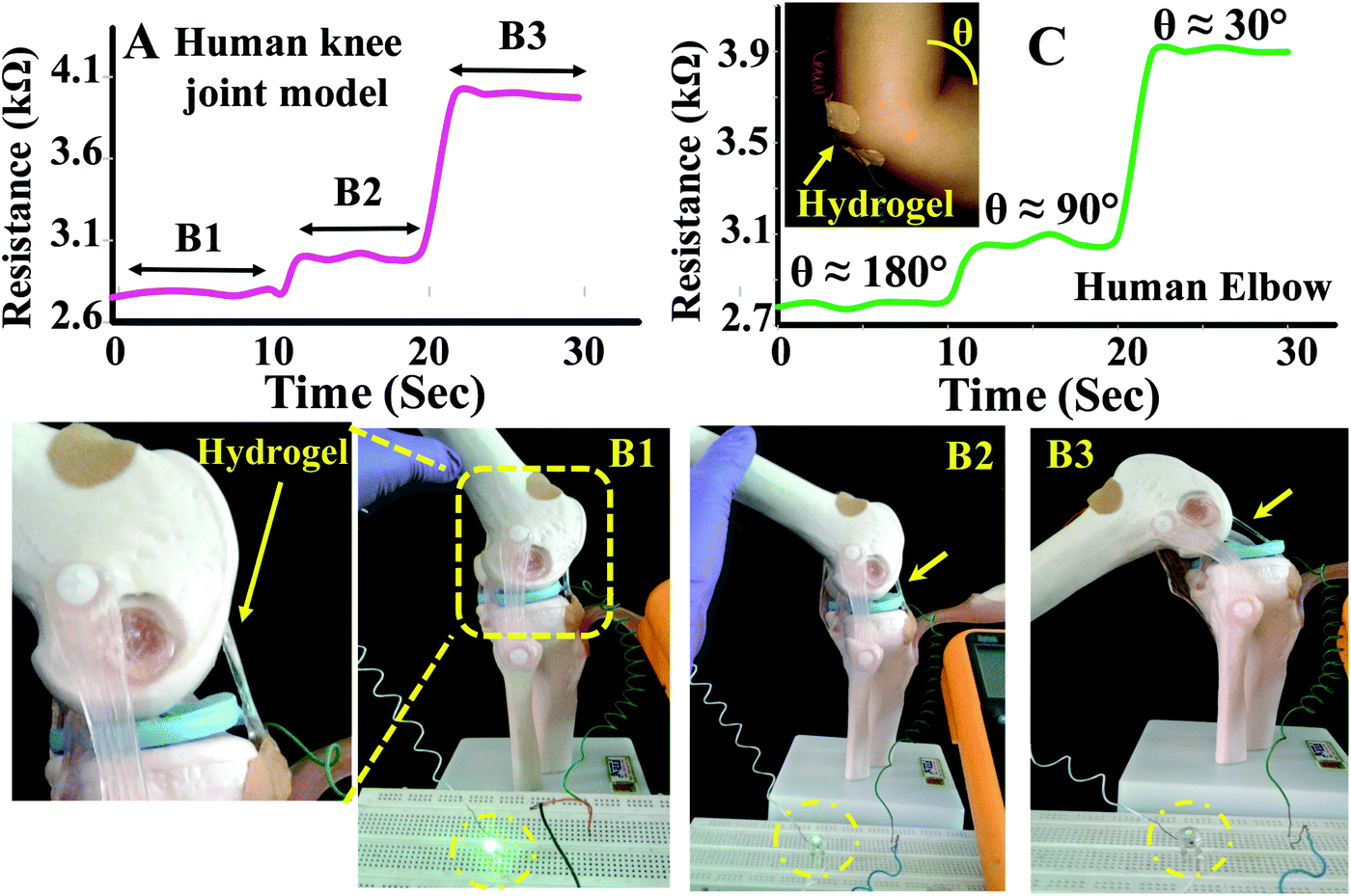 | ||
| Fig. 9 The change in resistance of a PAMS-PAH-PEG sample with movement of the (A) knee joint model and (C) elbow model. (B1–B3) Photographs showing the movements of a knee joint model attached with a PAMS-PAH-PEG strip at the joint. | ||
Similarly, the resistance of the sample changed from 2.7 to 3.9 kΩ with the movement of the elbow. The elbow and knee models were held at each position for 10 s to understand the fluctuations associated with the reading. The change in resistance value at each position was minimal in both cases suggesting that these hydrogel samples may be utilized to fabricate devices for sensing body movements by implementing certain amplification tools.
To summarize, PAHz incorporation created energy-dissipating micro-domains in the hydrogel matrix and improved the overall mechanical properties of the ionic hydrogels. PAMS-PAH-PEG hydrogel exhibited excellent adhesive properties towards glass substrates along with quick self-healing and strain sensing ability. The adhesive strength of these hydrogel samples was maximum on a glass surface and comparable to several reported systems. Similarly, the strain sensing ability of these hydrogel systems with gauge factor value up to 0.7 may be suitable for various applications. The study proved that this strategy is general and may be used to develop various tough and self-healable ionic hydrogels in the future.
Conclusion
PAHz capable of interacting with a polymerizable monomer through ionic interactions may be incorporated in small molar fractions to prepare ionic hydrogels with improved mechanical properties. These PAHz chains form possible network-reinforced zones in the hydrogel matrix that absorb stress through reversible rupture of the ionic bond and enhance the mechanical properties of the hydrogels. The strategy is successful in inducing autonomous self-healing ability, and adhesive and strain sensing properties in the hydrogels. The approach is general, and easy to implement and can be utilized for large scale synthesis of hydrogels using a simple one-pot procedure. These PAHz incorporated PAMPS-based hydrogels retain excellent elasticity and exhibit ultrahigh compressive strength value at low water content suggesting that these materials may be used as oleophobic rubbers. The hydrogels exhibit useful adhesive strength on glass surfaces, and strain sensing properties highlighting their usefulness for various applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support from DST-SERB (EMR/2016/006464), India is gratefully acknowledged.References
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, ACS Macro Lett., 2018, 7, 1105–1110 CrossRef CAS.
- M. J. Rowland, C. C. Parkins, J. H. McAbee, A. K. Kolb, R. Hein, X. J. Loh, C. Watts and O. A. Scherman, Biomaterials, 2018, 179, 199–208 CrossRef CAS PubMed.
- Z. Wang, F. Tao and Q. Pan, J. Mater. Chem. A, 2016, 4, 17732–17739 RSC.
- S. Hong, D. Sycks, H. F. Chan, S. Lin, G. P. Lopez, F. G. Kam, W. Leong and X. Zhao, Adv. Mater., 2015, 27, 4035–4040 CrossRef CAS PubMed.
- J. Liu, D. Su, J. Yao, Y. Huang, Z. Shao and X. Chen, J. Mater. Chem. A, 2017, 5, 4163–4171 RSC.
- S. Das, P. Chakraborty, A. Shit, S. Mondal and A. K. Nandi, J. Mater. Chem. A, 2016, 4, 4194–4210 RSC.
- J. Liu and O. A. Scherman, Adv. Funct. Mater., 2018, 28, 1800848 CrossRef.
- L. Chen, Y. Yin, Y. Liu, L. Lin and M. Liu, Chin. J. Polym. Sci., 2017, 35, 1181–1193 CrossRef CAS.
- P. M. Kharkar, R. A. Scott, L. P. Olney, P. J. LeValley, E. M. Kristi, L. Kiick and A. M. Kloxin, Adv. Healthcare Mater., 2017, 24, 1700713 CrossRef PubMed.
- Y. Liang, H. Wang, D. Yao, Y. Chen, Y. Deng and C. Wang, J. Mater. Chem. A, 2017, 5, 18442–18447 RSC.
- X. Xu, C. Heath, B. Pejcic and C. D. Wood, J. Mater. Chem. A, 2018, 6, 4829–4838 RSC.
- S. Das, P. Chakraborty, S. Mondal, A. Shit and A. K. Nandi, ACS Appl. Mater. Interfaces, 2016, 8, 28055–28067 CrossRef CAS PubMed.
- Q. Rong, W. Lei and M. Liu, Chem. – Eur. J., 2018, 24, 16930–16943 CrossRef CAS PubMed.
- A. M. Rosales, S. L. Vega, F. W. DelRio, J. A. Burdick and K. S. Anseth, Angew. Chem., Int. Ed., 2017, 56, 12132–12136 CrossRef CAS PubMed.
- Q. Rong, W. Lei, L. Chen, Y. Yin, J. Zhou and M. Liu, Angew. Chem., Int. Ed., 2017, 56, 14159–14163 CrossRef CAS PubMed.
- Q. Zhang, B. Chen, L. Tao, M. Yan, L. Chen and Y. Wei, RSC Adv., 2014, 4, 32475–32481 RSC.
- L. Chen, Z. Gu, L. Li, W. Lei, Q. Rong, C. Zhao, Q. Wu, Z. Gu, X. Jin, L. Jiang and M. Liu, J. Mater. Chem. A, 2018, 6, 15147–15153 RSC.
- X. Tong, L. Du and Q. Xu, J. Mater. Chem. A, 2018, 6, 3091–3099 RSC.
- C. B. Rodell, N. N. Dusaj, C. B. Highley and J. A. Burdick, Adv. Mater., 2016, 28, 8419–8424 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010 CrossRef PubMed.
- U. Haldar, M. Nandi, B. Maiti and P. De, Polym. Chem., 2015, 6, 5077–5085 RSC.
- Y. Yang, X. Wang, F. Yang, H. Shen and D. Wu, Adv. Mater., 2016, 28, 7178 CrossRef CAS PubMed.
- R. Takahashi, T. L. Sun, Y. Saruwatari, T. Kurokawa, D. R. King and J. P. Gong, Adv. Mater., 2018, 30, 1706885 CrossRef PubMed.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS PubMed.
- L. Jiang, C. Liu, K. Mayumi, K. Kato, H. Yokoyama and K. Ito, Chem. Mater., 2018, 30, 5013–5019 CrossRef CAS.
- L. W. Xia, R. Xie, X. J. Ju, W. Wang, Q. Chen and L. Y. Chu, Nat. Commun., 2013, 4, 2226 CrossRef PubMed.
- Z. Zhu, S. Ling, J. Yeo, S. Zhao, L. Tozzi, M. J. Buehler, F. Omenetto, C. Li and D. L. Kaplan, Adv. Funct. Mater., 2018, 28, 1704757 CrossRef.
- F. Chen, Q. Chen, L. Zhu, Z. Tang, Q. Li, G. Qin, J. Yang, Y. Zhang, B. Ren and J. Zheng, Chem. Mater., 2018, 30, 1743–1754 CrossRef CAS.
- L. J. Macdougall, V. X. Truong and A. P. Dove, ACS Macro Lett., 2017, 6, 93–97 CrossRef CAS.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722–2727 CrossRef CAS PubMed.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- Q. Chen, X. Yan, L. Zhu, H. Chen, B. Jiang, D. Wei, L. Huang, J. Yang, B. Liu and J. Zheng, Chem. Mater., 2016, 28, 5710–5720 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- M. A. Gonzalez, J. R. Simon, A. Ghoorchian, Z. Scholl, S. Lin, M. Rubinstein, P. Marszalek, A. Chilkoti, G. P. López and X. Zhao, Adv. Mater., 2017, 29, 1604743 CrossRef PubMed.
- X. Li, H. Wang, D. Li, S. Long, G. Zhang and Z. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 31198–31207 CrossRef CAS PubMed.
- J. You, S. Xie, J. Cao, H. Ge, M. Xu, L. Zhang and J. Zhou, Macromolecules, 2016, 49, 1049–1059 CrossRef CAS.
- S. Debnath, R. R. Ujjwal and U. Ojha, Macromolecules, 2018, 51, 9961–9973 CrossRef CAS.
- R. L. Bartlett, M. R. Medow, A. Panitch and B. Seal, Biomacromolecules, 2012, 13, 1204–1211 CrossRef CAS PubMed.
- Q. Zhang, N. Liu, Y. Wei and L. Feng, Soft Matter, 2018, 14, 2649–2654 RSC.
- B. A. Getachew, S. R. Kim and J. H. Kim, Environ. Sci. Technol., 2017, 51, 905–913 CrossRef CAS PubMed.
- J. Liang, G. Shan and P. Pan, Soft Matter, 2017, 13, 4148–4158 RSC.
- A. Kumar, R. R. Ujjwal, A. Mittal, A. Bansal and U. Ojha, ACS Appl. Mater. Interfaces, 2014, 6, 1855–1865 CrossRef CAS PubMed.
- R. R. Ujjwal, C. Sona, S. Debnath, P. N. Yadav and U. Ojha, ACS Omega, 2017, 2, 4278–4286 CrossRef CAS PubMed.
- A. H. Milani, L. A. Fielding, P. Greensmith, B. R. Saunders, D. J. Adlam, A. J. Freemont, J. A. Hoyland, N. W. Hodson, M. A. Elsawy, A. F. Miller, L. P. D. Ratcliffe, O. O. Mykhaylyk and S. P. Armes, Chem. Mater., 2017, 29, 3100–3110 CrossRef CAS.
- S. A. Zawko, S. Suri, Q. Truong and C. E. Schmidt, Acta Biomater., 2009, 5, 14–22 CrossRef CAS PubMed.
- J. Kim, J. A. Hanna, M. Byun, C. D. Santangelo and R. C. Hayward, Science, 2012, 335, 1201–1205 CrossRef CAS PubMed.
- X. Zhao, Proc. Natl. Acad. Sci. U. S. A. 2017, 201710942.
- R. E. Webber, C. Creton, H. R. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919–2927 CrossRef CAS.
- W. J. Zheng, N. An, J. H. Yang, J. Zhou and Y. M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 1758–1764 CrossRef CAS PubMed.
- E. Hoenig, U. Leicht, T. Winkler, G. Mielke, K. Beck, F. Peters, A. F. Schilling and M. M. Morlock, Tissue Eng., Part A, 2013, 19, 1534–1542 CrossRef CAS PubMed.
- X. Liang and S. A. Boppart, IEEE Trans. Biomed. Eng., 2010, 57, 953–959 Search PubMed.
- M. Zhong, Y. T. Liu and X. M. Xie, J. Mater. Chem. B, 2015, 3, 4001–4008 RSC.
- F. K. Shi, M. Zhong, L. Q. Zhang, X. Y. Liu and X. M. Xie, J. Mater. Chem. B, 2016, 4, 6221–6227 RSC.
- F. Yang, V. Tadepalli and B. J. Wiley, ACS Biomater. Sci. Eng., 2017, 3, 863–869 CrossRef CAS.
- S. Liu and L. Li, ACS Appl. Mater. Interfaces, 2016, 8, 29749–29758 CrossRef CAS PubMed.
- X. Li, Q. Yang, Y. Zhao, S. Long and J. Zheng, Soft Matter, 2017, 13, 911–920 RSC.
- J. C. McKeen, Y. S. Yan and M. E. Davis, Chem. Mater., 2008, 20, 3791–3793 CrossRef CAS.
- S. Yusa, Y. Yokoyama and Y. Morishima, Macromolecules, 2009, 42, 376–383 CrossRef CAS.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- D. L. Taylor and M. Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- R. Tian, X. Qiu, P. Yuan, K. Lei, L. Wang, Y. Bai, S. Liu and X. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 17018–17027 CrossRef CAS PubMed.
- H. Gong, Y. Gao, S. Jiang and F. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 26694–26704 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- A. Li, Y. Jia, S. Sun, Y. Xu, B. B. Minsky, C. M. A. Stuart, H. Colfen, R. Klitzing and X. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 10471–10479 CrossRef CAS PubMed.
- V. Bhagat and M. L. Becker, Biomacromolecules, 2017, 18, 3009–3039 CrossRef CAS PubMed.
- H. Fan, J. Wang, Q. Zhang and Z. Jin, ACS Omega, 2017, 2, 6668–6676 CrossRef CAS PubMed.
- J. Newar and A. Ghatak, Langmuir, 2015, 31, 12155–12160 CrossRef CAS PubMed.
- C. Shao, M. Wang, L. Meng, H. Chang, B. Wang, F. Xu, J. Yang and P. Wan, Chem. Mater., 2018, 30, 3110–3121 CrossRef CAS.
- M. Shan, C. Gong, B. Li and G. Wu, Polym. Chem., 2017, 8, 2997–3005 RSC.
- X. Jing, H. Y. Mi, Y. J. Lin, E. Enriquez, X. F. Peng and L. S. Turng, ACS Appl. Mater. Interfaces, 2018, 10, 20897–20909 CrossRef CAS PubMed.
- L. Han, X. Lu, K. Liu, K. Wang, L. Fang, L. T. Weng, H. Zhang, H. Tang, F. Ren, C. Zhao, G. Sun, R. Liang and Z. Li, ACS Nano, 2017, 11, 2561–2574 CrossRef CAS PubMed.
- P. Rao, T. L. Sun, L. Chen, R. Takahashi, G. Shinohara, H. Guo, D. R. King, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1801884 CrossRef PubMed.
- S. Liu and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 26429–26437 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Schematics of experimental set-up, swelled hydrogel photographs, FESEM, hysteresis and tensile data. See DOI: 10.1039/c8qm00659h |
| ‡ SM and NP contributed equally to the work. |
| This journal is © the Partner Organisations 2019 |