
All-inorganic lead-free perovskites for optoelectronic applications
Xingtao
Wang
a,
Taiyang
Zhang
a,
Yongbing
Lou
 *b and
Yixin
Zhao
*a
*b and
Yixin
Zhao
*a
aSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: yixin.zhao@sjtu.edu.cn
bSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing, 211189, China. E-mail: lou@seu.edu.cn
First published on 28th January 2019
Abstract
Organic and inorganic hybrid lead halide perovskites have successfully emerged as revolutionary optoelectronic semiconductors for use in various applications. The long-term stability and lead toxicity of hybrid lead halide perovskites have attracted increased attention; therefore, all-inorganic lead-free perovskites have become an alternative perovskite for use in optoelectronic applications. Among them, all-inorganic CsSnI3 has also been developed. The main issue limiting the optoelectronic performance and stability of Sn-based perovskites is the low chemical stability of Sn2+, which is very easy to be oxidized to Sn4+ under air conditions. Several approaches have been adopted to prevent the oxidation of Sn, thereby improving its performance. However, its chemical stability is still difficult to manage. Other than Sn, other transition metals such as Ge, Bi, and Cu have also been used to substitute Pb and form novel lead-free perovskites. Although such non-Sn lead-free perovskites exhibit much better stability, their photovoltaic performances are lower as compared to those of Pb- or Sn-based perovskites. However, these novel all-inorganic lead-free perovskites exhibit potential in photoluminescence and other optoelectronic applications. Overall, we have reviewed the recent progresses and outlooks regarding the prospects and challenges faced by all-inorganic lead-free perovskites.
 Yongbing Lou | Prof. Yongbing Lou received his BS degree from Nankai University in 1996 and received his MS degree from Nanjing University in Polymer Materials in 1999. After acquiring his PhD in Chemistry from Case Western Reserve University in 2007, he joined the School of Chemistry and Chemical Engineering at Southeast University. His research interests include the preparation of novel nanomaterials for optoelectronic, sensing, and energy-related applications. He has published more than 50 peer-reviewed papers. |
 Yixin Zhao | Yixin Zhao is a professor at Shanghai Jiao Tong University. He obtained his PhD from Case Western Reserve University in 2010, followed by working as a postdoctoral fellow at Penn State University and National Renewable Energy Laboratory. His current research interests focus on perovskite solar cells, photoelectrochemical catalysis, and environmental remediation. Professor Zhao is on the editorial board for several journals and has coauthored over 110 peer-reviewed publications. |
1. Introduction
Organic–inorganic lead halide perovskites (generally having the formula ABX3, where A is a monovalent organic cation, B is a lead cation, and X is a halide anion) have emerged as one of the most promising potential optoelectronic materials in the past few years because of their tunable direct bandgap, high absorption coefficient, low exciton binding energy, and high carrier mobility.1–5 Various applications including solar cells, light-emitting devices, photocatalysis, photodetectors, and lasers have been reported.6–13 The power conversion efficiency (PCE) of solar cells formed using halide perovskites have been improved from the initially unstable 3.8% in 2009 to the recently certified 23.3%.14–23 In spite of the considerable progress made in the fabrication of hybrid lead halide perovskite materials, there are still some scientific challenges and technical obstacles for their commercial applications.24 Among them, lead toxicity is a fatal drawback of using hybrid lead halide perovskites. Therefore, it is imperative to develop alternative lead-free perovskites with excellent performance that can be used in optoelectronic applications.It has been reported that lead's lone pair 6s2 electrons that align with the halides to form antibonding coupling play an important role in the excellent photovoltaic properties of lead halide perovskites.25–27 Therefore, metal elements that have similar electronic structures as Pb to form stable perovskites have been considered as a suitable alternative. Sustained efforts have been devoted toward developing lead-free perovskites that have prominent photovoltaic performance.28,29 For example, it has been demonstrated that MASnI3 exhibited narrower bandgap than MAPbI3.30 The Pb–Sn alloyed perovskite is the main candidate for preparing low-bandgap perovskite solar cells. Noel et al. reported MASnI3 perovskites as 1.23 eV-based solar cells with PCE of over 6% under 1 illumination.31 Bi-based perovskites exhibited superior stability than their Pb analogues,32–34 indicating that Pb could be successfully substituted with other elements.
Since the organic cation may affect long-term photostability, alternative inorganic cations could further enhance the robustness of perovskite materials and devices.13,35–43 Therefore, there are still several challenges in developing more stable perovskite photoelectronic devices with higher efficiencies. In this review, we summarized the recent advances in all-inorganic lead-free perovskites and discussed various candidates and approaches to improve their optoelectronic performance.
2. Lead substitution
The Goldschmidt's tolerance factor (t) that depends on the ionic radius of the ions in perovskites is an empirical parameter that can be used to determine the structure of perovskites.where RA, RB, and RX are the ionic radii of A, B, and X ions, respectively.44 A stable perovskite structure can be predicted when t is in the range from 0.75 to 1.0.45 The ionic radius of lead is 1.19 Å.46 Therefore, lead in perovskites can be substituted by several alternative metal elements with similar ionic radii. Zhang and coworkers theoretically identified 14 lead-free perovskites based on Group 14 elements (Sn, Ge) and 11 nontoxic double perovskites with suitable bandgaps, intrinsic thermodynamic stabilities, small carrier effective masses, and low exciton binding energies by means of first-principles calculations.47,48 These candidates are shown in Fig. 1.
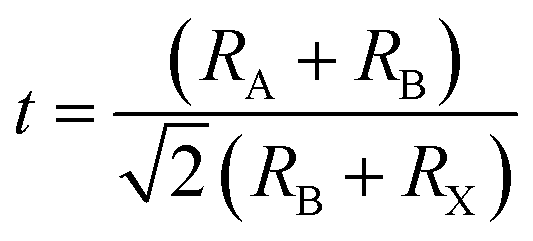
 | ||
| Fig. 1 Distribution of (a) AMIVXVII3 perovskites and (b) A2M+M3+XVII6 perovskites with respect to the octahedral factor (μ) and tolerance factor (t). Reprinted with permission from ref. 47 and 48. Copyright 2017 American Chemical Society. | ||
In this review, we summarize several comprehensively studied candidates for substitution with Pb. The most straightforward candidates are Group 14 elements, namely, Sn and Ge, which have electrical structures similar to that of Pb. Then, there are Group 15 elements, namely, Bi and Sb: trivalent Bi3+ and Sb3+ possess similar electronic configurations. As compared to Pb, they can exhibit more diverse dimensionalities due to the presence of the BiX63− (SbX63−) octahedron, and they are much less toxic than Pb. Recently, Cu and Ti have become new candidates, which are earth-abundant, nontoxic, and biocompatible elements. Table 1 lists the ionic radii and electronic configurations of metal cations for the substitution of Pb.
| Metal cations | Ionic radius (Å) | Electronic configuration |
|---|---|---|
| Pb2+ | 1.19 | 6s2 |
| Sn2+ | 1.02 | 5s2 |
| Ge2+ | 0.73 | 4s2 |
| Bi3+ | 1.03 | 6s2 |
| Sb3+ | 0.76 | 5s2 |
| Sn4+ | 0.69 | 4d10 |
| Ti4+ | 0.53 | 3p6 |
| Cu2+ | 0.73 | 3d9 |
2.1 Sn
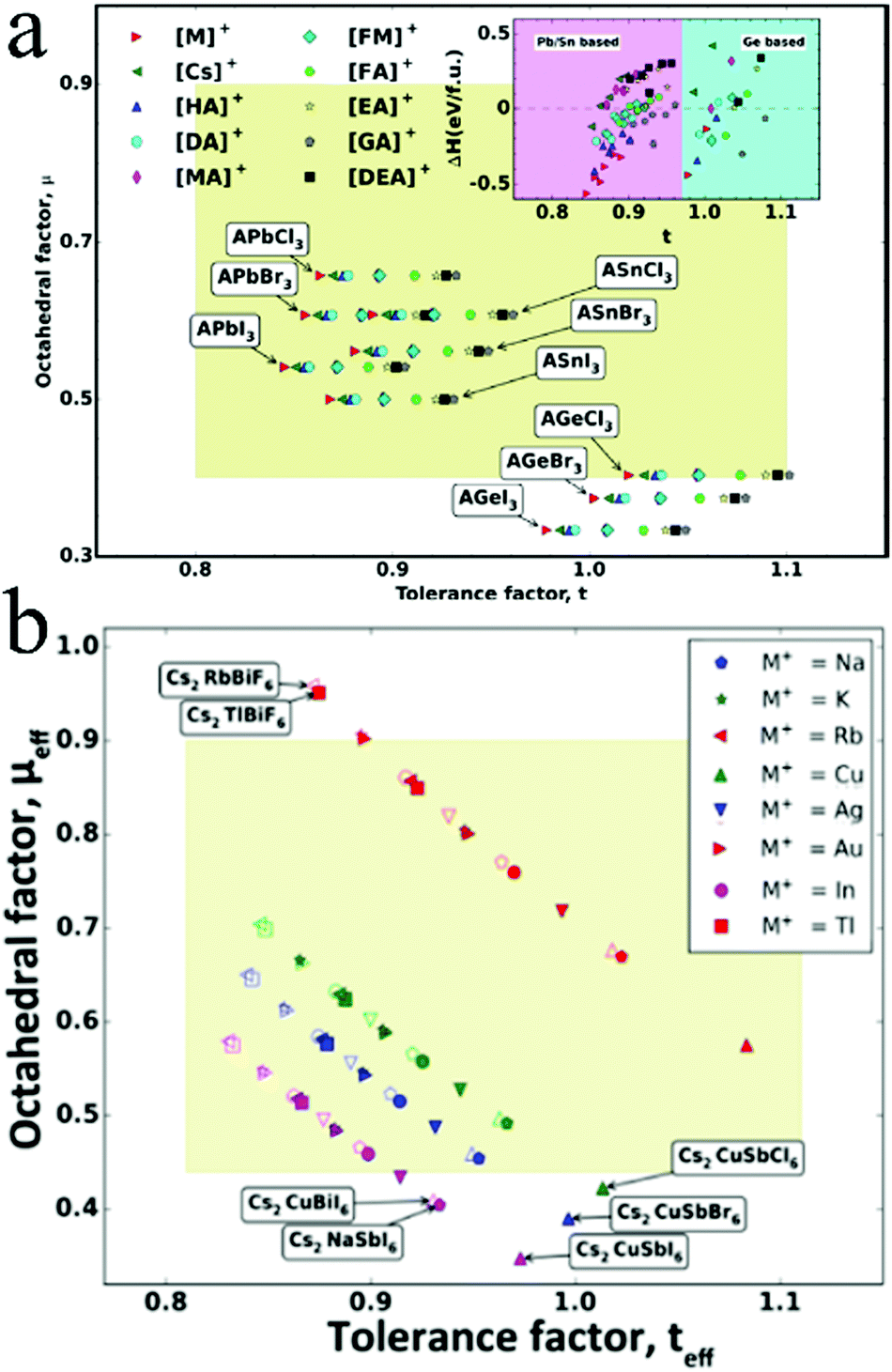
Sn, a Group 14 element with an electronic structure similar to lead, has been firstly considered as the alternative metal for halide perovskite materials. In addition, Sn-based perovskites exhibit a narrower bandgap than their Pb analogues with low exciton binding energies and longer carrier diffusion lengths.49,50 Therefore, Sn-based perovskites are regarded as promising light absorbers in optoelectronic applications. Considerable efforts have been devoted toward formulating Sn-based perovskites over the past few years. Snaith and coworkers reported lead-free MASnI3 perovskite solar cells with PCE of over 6%.31 Hao et al. reported that the band structure of MASnI3 could be tuned by substituting iodide with bromide to cover more visible-light region.30 Ma et al. suggested that the carrier diffusion length of MASnI3 could exceed 500 nm after doping SnF2, indicating the potential of MASnI3-based solar cells.51 Liu et al. combined hot antisolvent treatment and dimethyl sulfoxide (DMSO) vapor to increase the film coverage and average crystallite size of FA0.75MA0.25SnI3, achieving PCE exceeding 7%.52 Qian et al. fabricated 2D perovskite (PEA)2SnI4 in flexible photoconductors with excellent photoresponsivity of 16 A W−1 and detectivity of 1.92 × 1011 Jones under 470 nm illumination.53 However, Sn-based perovskites are sensitive toward moisture and oxygen owing to the oxidation process of Sn2+ to Sn4+. In addition, the product of Sn4+—a p-type dopant—makes the materials metallic via a self-doping process.54 This oxidation process results in instabilities and poor performance of optoelectronic devices. Various strategies have been developed to inhibit the oxidation of Sn2+, including adding additives in the precursor solution, adjusting the atmosphere, and controlling the morphology of the perovskite crystal.In order to further improve the Voc value of CsSnX3 materials, the impact of Br− replacing I− was exploited. Sabba et al. demonstrated that the substitution of I with Br is an effective way to improve the Voc value of CsSnI3 solar cells.58 The crystal structure of CsSnI3 changing from orthorhombic to cubic for CsSnBr3 was accompanied with the bandgap changing from 1.27 eV to 1.75 eV by progressively replacing I− with Br− (Fig. 2a). Br-Rich perovskites exhibited low carrier densities and fewer charge recombinations, resulting in high open-circuit voltages. With the increase in Br doping, Voc increased from 2.30 mV for CsSnI3 to 190 mV for CsSnBr3 even without the addition of SnF2.
 | ||
| Fig. 2 (a) Crystal structure and bandgap of CsSnX3 (X = I, Br). Reprinted with permission from ref. 58. Copyright 2015 American Chemical Society. (b) Stability of CsSnI3 with different Sn salt additives. Reprinted with permission from ref. 59. Copyright 2016 Springer Nature. (c) Possible mechanism of hydrazine preventing the oxidation of Sn2+. Reprinted with permission from ref. 54. Copyright 2017 American Chemical Society. | ||
The role of SnF2 on the properties of CsSnBr3 was also investigated.60 The ultraviolet photoelectron spectroscopy (UPS) data reveal that the conduction band minimum (CBM) of CsSnI3 perovskite increases with doping SnF2, which would reduce the electron injection barrier between the perovskite and CBM of the TiO2 layer. Therefore, CsSnBr3 with SnF2 doping exhibited fewer recombinations than those in pristine perovskites. Further, the addition of SnF2 moves the valence band maximum (VBM) of the perovskite closer to the HOMO level of HTM (spiro), thereby decreasing the large loss of voltage at the HTM–perovskite interface. Consequently, perovskite solar cell (PSC) devices based on CsSnBr3 with 20 mol% SnF2 exhibited enhanced photovoltaic performance with η = 2.17%, Jsc = 9.1 mA cm−2, Voc = 0.42 V, and FF = 0.56 as compared to pristine CsSnBr3 with η = 0.01%, Jsc = 0.4 mA cm−2, Voc = 0.1 V, and FF = 0.33. This result indicates that the performance of CsSnBr3-based devices can be improved by tuning the band alignment.
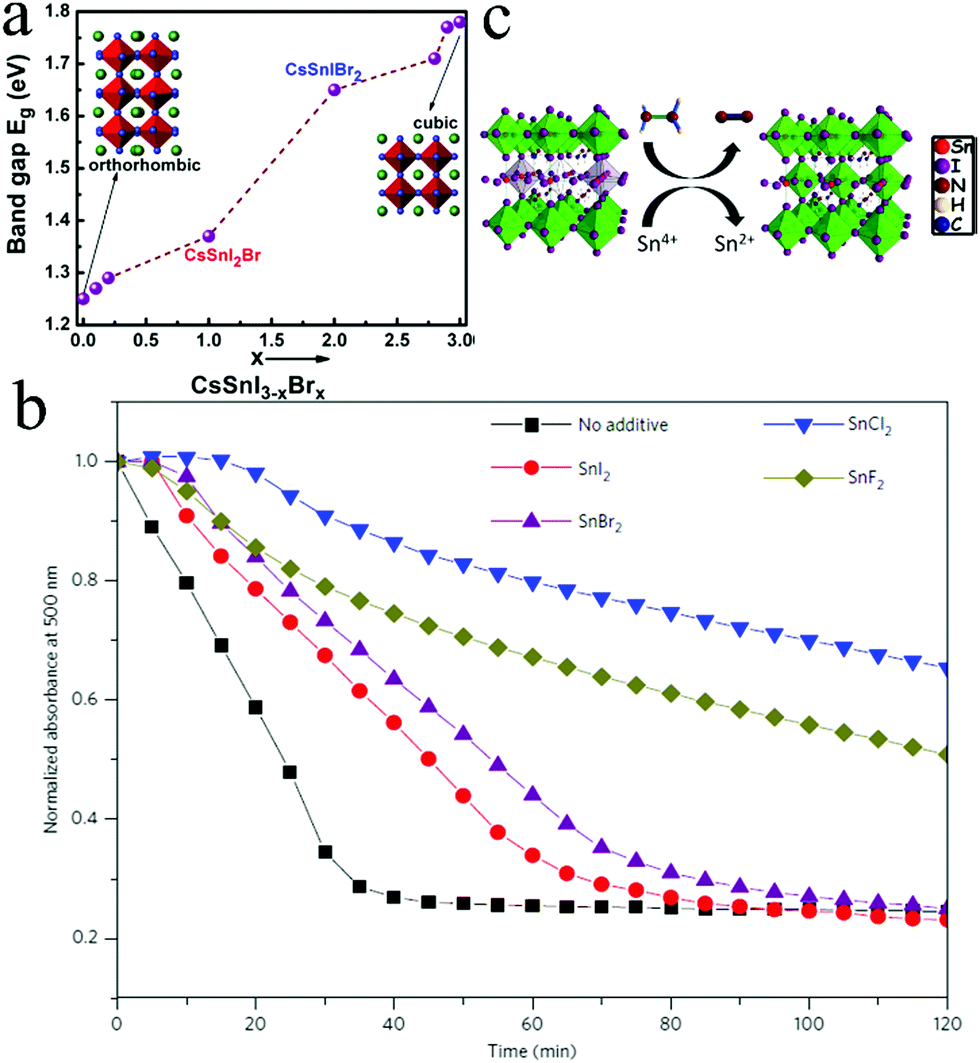
Other Sn halide salts such as SnI2, SnCl2, and SnBr2 were also introduced as additives to improve PCE and stability. The introduction of 10% excess SnI2 in CsSnI3 preparation increased the device performance from η = 0.75% to η = 1.5%.61 However, CsPbI3-based PSC devices are highly sensitive to air and their efficiencies quickly decreased more than 30% after storing in the dark in ambient air for 1 h. Further research revealed that excess SnCl2 could form a thin layer on the surface of CsSnI3 crystallites because Cl cannot substitute I in CsSnI3.59 However, the SnCl2 layer could protect CsSnI3 by forming a stable hydrate (SnCl2·2H2O), resulting in better stability than other Sn salt additives (Fig. 2b). In addition, SnCl2 n-doped the fullerene layer, allowing the device to function effectively without an electron blocking layer (EBL): this minimized the pinhole density of the film. Therefore, devices using SnCl2 as additives exhibited the highest η value due to a simple fabricating process with η = 3.26%, Jsc = 11.25 mA cm−2, Voc = 0.40 V, and FF = 0.55.
The effect of SnBr2 as the additive in CsSnBr3 was investigated via the vapor deposition method using CsBr and excess SnBr2.62 The Voc value of CsSnBr3-based devices without SnF2 was enhanced because excess SnBr2 reduced the density of the defect states and prevented the oxidation of Sn2+ to Sn4+ states in ambient air. This demonstrated that SnF2 dopants passivated bulk and grain boundary defects. Therefore, devices based on CsSnBr3 with 2.5% mol SnF2 retained 80% of their performance over 50 min, and the films retained the same absorption value over 24 h; however, CsSnI3 and CsSnI2Br degraded within a few minutes when exposed to air. This indicated that CsSnBr3 could be a promising air-stable all-inorganic lead-free perovskite material as compared to CsSnI3. Besides Sn halide salts, hypophosphorous acid (HPA) was also introduced as an additive in CsSnIBr2.63 HPA could strongly coordinate with Sn2+ and stabilize the CsSnIBr2 precursor by generating Sn–O–P–O–Sn bond connection, preventing the oxidation of Sn2+. The amount of Sn4+ in CsSnIBr2 with HPA was much lower than that of the pristine film. Further, the introduction of HPA did not induce phase impurity. In addition, HPA reduced the carrier mobility and charge carrier density of the CsSnIBr2 film, which inhibited charge recombinations. Therefore, devices based on HPA addition achieved average PCE of 3.0% with Jsc = 17.4 mA cm−2, Voc = 0.31 V, and FF = 0.56. Furthermore, devices retained 98% of their performance up to 473 K.
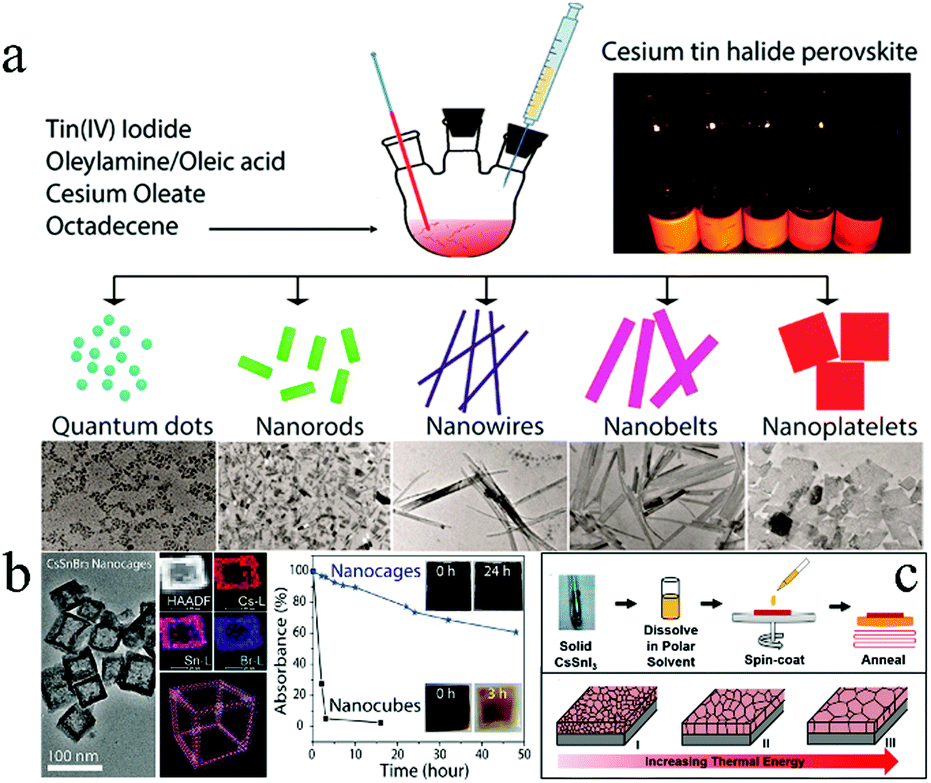 | ||
| Fig. 3 (a) Cs2SnX6 (X = Cl, Br, I) with different morphologies. Reprinted with permission from ref. 66. Copyright 2016 American Chemical Society. (b) Morphology and stability of CsSnBr3 nanocages. Reprinted with permission from ref. 67. Copyright 2017 American Chemical Society. (c) Grain coarsened with an increase in temperature. Reproduced with permission from ref. 68. Copyright 2016 John Wiley and Sons. | ||
In order to improve stability, hollow CsSnBr3 nanocages (Fig. 3b) were synthesized by a hot-injection colloidal approach for further advancing Sn-based perovskites.67 Hollow nanocages showed enhanced stability than perovskites with other morphologies under ambient conditions (Fig. 3b). In addition, NCs were treated with perfluorooctanoic acid (PFOA) to passivate the surface of the CsSnBr3 nanocage, which significantly improved its stability against moisture, oxygen, and light. Uniform CsSnX3 (X = Cl, Br, I) quantum rods (QRs) were successfully synthesized by a solvothermal method.70 These QRs exhibited tunable emission wavelengths ranging from 625 to 709 nm that could be achieved by adjusting the halide composition. The maximum external quantum efficiency (EQE) peaks of these QRs reached over 63% (CsSnCl3), 65% (CsSnBr3), and 70% (CsSnI3) because of the larger surface area of QRs as compared to the bulk materials. Furthermore, the stability of CsSnI3 was enhanced as compared to MAPbI3. Further, it was reported that the grain morphology was an important factor affecting the performance of the devices.68 Abundant grain boundaries in the thin films resulted in a significant recombination of electrons–holes. The grain size of the CsSnI3 thin film increased with fewer grain boundaries after high-temperature annealing (Fig. 3c). In addition, NiOx was introduced as the hole transport material for matching the energy level of CsSnI3. PCE of 3.31% with Voc = 0.52 V, Jsc = 10.21 mA cm−2, and FF = 0.625 could be achieved under optimized conditions.
2.2 Ge
Ge, another Group 14 element, was also considered as a potential candidate for Pb substitution in halide perovskites. The structural and electronic properties of MAGeX3 (X = Cl, Br, I) were investigated by theoretical calculations.74 The results revealed that MAGeI3-based perovskites exhibited similar hole and electron conductive behaviors, stabilities, and optical properties as those of MAPbI3. This indicated that Ge was a competitive alternative to Pb for use in perovskites. AGeI3 (A = MA, FA, Cs) was prepared by Krishnamoorthy et al.75Fig. 4a shows the band structure of three perovskites. Among them, CsGeI3 (Fig. 4b) exhibited the highest thermal stability (up to 350 °C) when compared with the other two Ge-based perovskites.76 However, solar cells exhibited poor performance due to the oxidation of Ge2+. CsGeX3 (X = Cl, Br, I) QRs (Fig. 4c) were prepared by a solvothermal method.77 The emission wavelength could be tuned from 607 to 696 nm by changing the composition of the different halides (Fig. 4d). PCE of 4.94% for solar cells based on CsGeX3 could be achieved with Voc = 0.51 V, FF = 0.51, and Jsc = 18.78 mA cm−2. The EQE values of the top-performing devices were observed to exceed 66% (for CsGeCl3), 68% (for CsGeBr3), and 79% (for CsGeI3).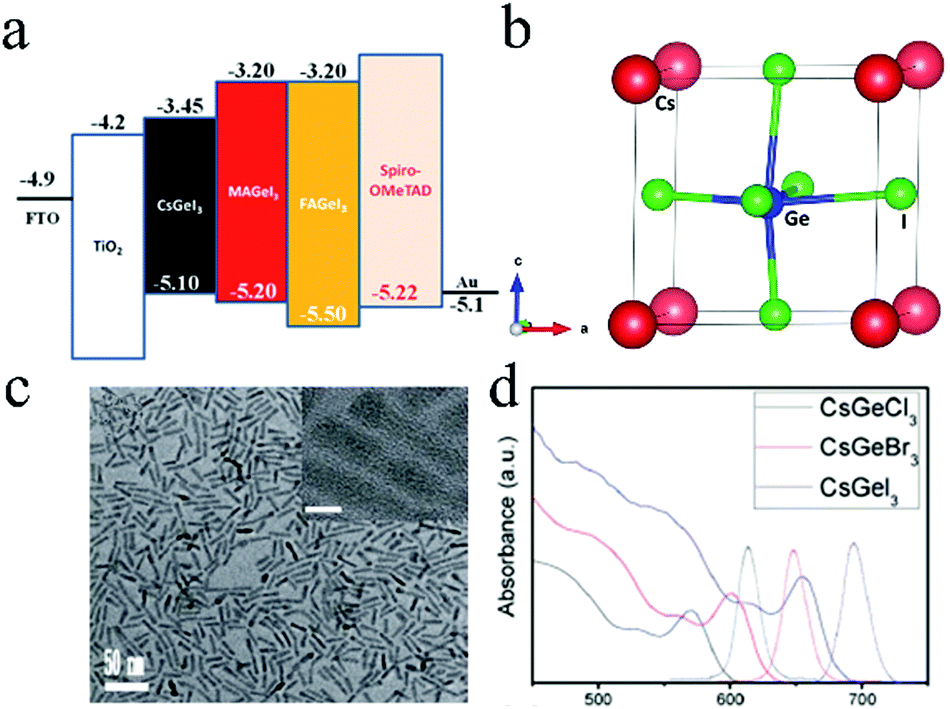 | ||
| Fig. 4 (a) Band structure of CsGeI3, MAGeI3, and FAGeI3. Reproduced with permission from ref. 75. Copyright 2015 The Royal Society of Chemistry. (b) Crystal structure of CsGeI3. Reproduced with permission from ref. 76. Copyright 2016 The Royal Society of Chemistry. (c) Morphologies of CsGeX3 (X = Cl, Br, I) QRs and (d) absorption spectra of CsGeX3 (X = Cl, Br, I). Reproduced with permission from ref. 77. Copyright 2018 The Royal Society of Chemistry. | ||
2.3 Bi
Bi is considered as a potential substitute because its valence electronic shell is isoelectronic to Pb. Furthermore, its ionic radius (1.03 Å) is similar to that of Pb (1.19 Å).78 Therefore, Bi has attracted considerable attention for use in halide perovskite optical applications. In the structure of A3Bi2X9, A and X atoms are the closest-packed and Bi atoms occupy two-thirds of the octahedral X6 voids. Park et al. prepared Bi-based perovskites with the chemical structure A3Bi2I9 (A = MA+, Cs+), which consisted of (Bi2I9)3 clusters surrounded by cations (Fig. 5a).79 Cs3Bi2I9 exhibited five times higher PL intensity when compared with the other two perovskites (MA3Bi2I9Clx and MA3Bi2I9). Cs3Bi2I9-based devices exhibited the best performance with Jsc = 2.15 mA cm−1, Voc = 0.85 V, FF = 0.60, and PCE = 1.09% (Fig. 5b). Furthermore, the light absorption spectra and PCE showed marginal changes after one-month storage (in dry air, humidity less than 10%, and in the dark). It has been demonstrated that Bi-based perovskites exhibited high PLQY and stability against moisture and heat. Such enhanced stabilities could be attributed to their lower-dimensional crystal phase as compared to regular 3D Pb halide perovskites, but the lower dimension also accounts for the lower PV performance. Cs3Bi2X9 NCs were synthesized by a facile room-temperature reaction.80 The emission wavelength could be tuned from 400 nm to 560 nm with different halide compositions. The PLQY of Cs3Bi2Br3 was improved from 0.2% to 4.5% by adding oleic acid surfactant that passivated the fast trapping process. Furthermore, Cs3Bi2Br9 NCs exhibited high air stability for over 30 days because the adsorption of water on the surface of Cs3Bi2Br9 formed perovskite hydrates, thereby passivating the perovskite.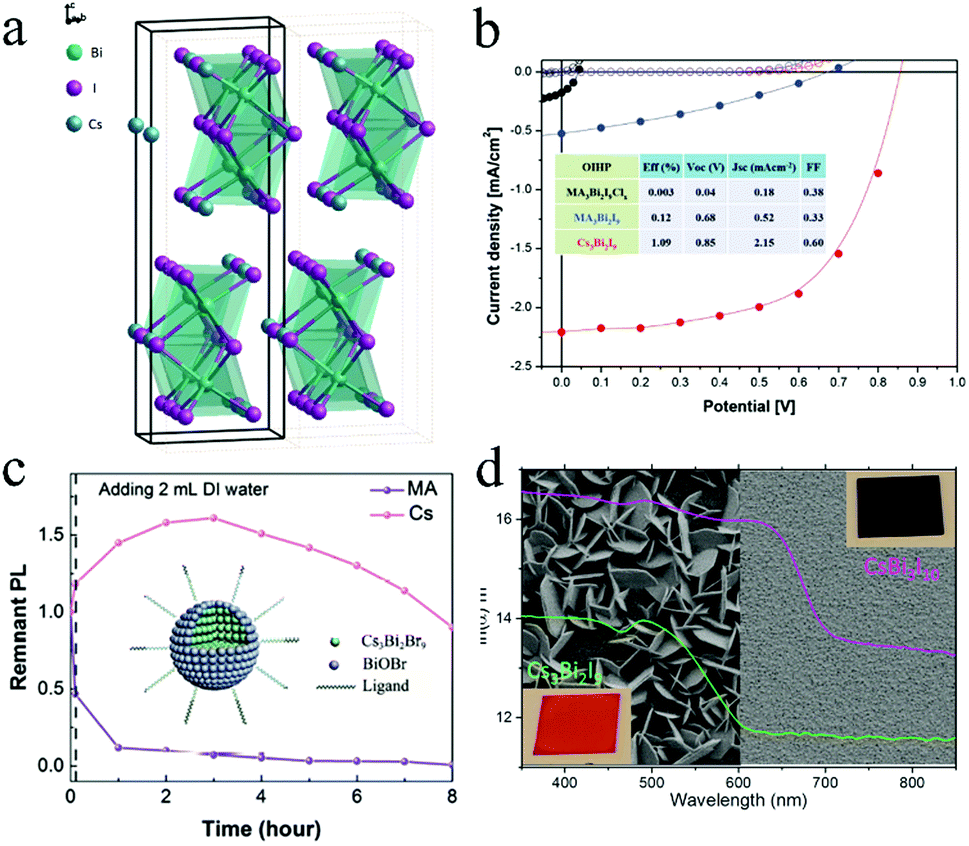 | ||
| Fig. 5 (a) Crystal structure of Cs3Bi2I9 and (b) performance of solar cells fabricated using Bi-based perovskites. Reprinted with permission from ref. 79. Copyright 2015 John Wiley and Sons. (c) Stability of Cs3Bi2Br9 against moisture.33 (d) Quality of Cs3Bi2I9 and CsBi3I10 films. Reprinted with permission from ref. 81. Copyright 2016 American Chemical Society. | ||
Lou and coworkers reported Cs3Bi2X9 (X = Cl, Br, I) QDs prepared by the steady binding of octylammonium halide (OA–X) and oleic acid.82 Cs3Bi2Br9 exhibited PLQY of up to 22% with an emission peak at 414 nm and a full width at half maximum value of 38 nm, which could remain stable even up to 180 °C. In addition, Cs3Bi2Cl9 and Cs3Bi2I9 could be easily obtained by a fast-reversible anion exchange reaction, exhibiting fluorescence emission wavelengths ranging from 380 nm to 526 nm with PLQYs of 62% and 2.3%, respectively.
Nelson et al. synthesized Cs3Bi2Br9 NCs by a hot-injection approach.83 The growth of Cs3Bi2Br9 was initiated by the formation of Bi0 seeds, and the morphologies were controlled by the precursor and seed concentrations. The perovskites exhibited excellent stability with unchanged characteristic absorbance peak at 489 nm exposed to the ambient condition. Cs3Bi2X9 perovskite QDs were reported by Leng et al. using ethanol as the main solvent.33 The tunable PL peaks of the perovskites were adjusted from 393 nm to 545 nm by means of different halide compositions. Cs3Bi2Br9 QDs achieved PLQY of ∼19.4% with the emission peak at 410 nm and FWHM value of 48 nm. Furthermore, as shown in Fig. 5c, the hydrolytic product BiOBr passivated the surface defects of the perovskite when Cs3Bi2Br9 was exposed to the ambient condition, resulting in good stability. Cs3Bi2Br9 QDs exhibited only 10% relative reduction in PL intensity after storage for 8 h.
Johansson et al. synthesized CsBi3I10 with a smaller bandgap (1.77 eV) than that of Cs3Bi2I9.81 As shown in Fig. 5d, CsBi3I10 exhibited a layered crystal structure, giving more uniform coating, while Cs3Bi2I9 dominated in a different growth direction. The solar cells based on CsBi3I10 yielded higher photocurrent, which could be attributed to the extended light harvesting originating from the smaller bandgap as compared to that of Cs3Bi2I9. However, the PCE of devices was still low (0.4%). The CsBi3I10 film was stable in ambient air for 17 h.
2.4 Sb
Trivalent Sb with lone-pair 5s2 electrons was considered as another candidate for lead substitution.84,85 Furthermore, Sb is more than twice as cheap as Sn.86 The structure of A3Sb2X9 is the same as A3Bi2X9, where Cs3Sb2I9 also possessed the 0D dimer form and 2D layered form. (NH4)3Sb2IxBr9−x (0 ≤ x ≤ 9) was prepared using ethanol as the solvent. The absorption peak could be tuned from 558 nm to 453 nm with an increase in Br. Although the PCE of (NH4)3Sb2I9-based devices was still low (0.51%), this work provided a nontoxic way to fabricate perovskite solar cells.87 The all-inorganic perovskite Cs3Sb2I9 was prepared by a two-step deposition approach.88Fig. 6a shows the 2D structure of Cs3Sb2I9. Cs3Sb2I9 exhibited indirect bandgap of 2.05 eV and enhanced stability against ambient conditions as compared to those of MAPbI3. However, the solar cells based on Cs3Sb2I9 exhibited a Voc value of 0.25–0.30 eV, which is lower than its organic–inorganic hybrid analogue (CH3NH3)3Sb2I9 (0.89 V).89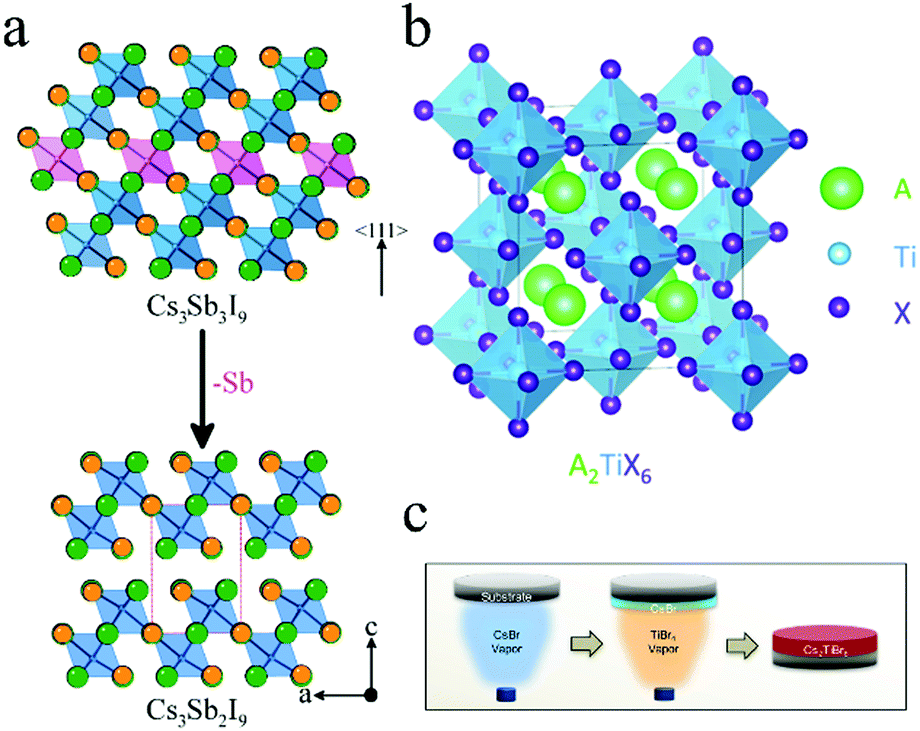 | ||
| Fig. 6 (a) Structures of Cs3Sb3I9 and Cs3Sb2I9. Reprinted with permission from ref. 88. Copyright 2015 American Chemical Society. (b) Structure of A2TiX6. Reprinted with permission from ref. 90. Copyright 2018 American Chemical Society. (c) Schematic illustration for preparing Cs2TiBr6 by vapor deposition method. Reprinted with permission from ref. 91. Copyright 2018 Elsevier. | ||
2.5 Ti
Ti, an earth-abundant, nontoxic, and ultrastable element, was discovered as a promising candidate for substitution with Pb.90Fig. 6b shows the crystal structure of A2TiX6. As compared to APbX3, A2TiX6 has a vacancy-ordered double perovskite structure, similar to that of A2SnX6. Padture and coworkers prepared Cs2TiIxBr6−x and its bandgap could be tuned from 1.02 eV to 1.78 eV.90 This group also synthesized Cs2TiBr6 by a two-step vapor deposition method (Fig. 6c).91 Cs2TiBr6 thin films showed bandgap of 1.82 eV with carrier diffusion lengths for electrons and holes of 121 nm and 103 nm, respectively. C60 facilitated electrons transfer from Cs2TiBr6 to TiO2 and improved the microstructure of Cs2TiBr6 thin films. Therefore, solar cells based on Cs2TiBr6 achieved PCE of 3.28% with Voc = 1.02 V, FF = 0.56, and Jsc = 5.69 mA cm−2. Furthermore, Cs2TiBr6 thin films exhibited much higher tolerance against heat, moisture, and light when compared with MAPbI2Br.2.6 Cu
Cu, a transition metal with the electronic configuration 3d9 (t62ge3g), has attracted considerable attention due to the excellent stability of its oxidation state and large absorption coefficient of its compounds.92 The organic–inorganic hybrid copper-based perovskite C6H4NH2CuBr2I was prepared with excellent hydrophobic properties. The XRD patterns remained unchanged even after 4 h of water immersion. However, solar cells based on such perovskites exhibited poor photovoltaic performance with PCE of 0.5%.93 All-inorganic copper-based perovskite Cs2CuX4 (X = Cl, Br, Br/I) QDs were firstly synthesized by Lou and coworkers.94 PL peaks could be tuned from 385 nm to 468 nm by the anion exchange reaction. Furthermore, the bandgap tuning of such perovskites could be attributed to the particle size by adjusting the ratios of the precursor. These copper-based perovskites exhibited excellent air and photostability when compared with other lead-free perovskites. Cs2CuBr4 maintained 92% of the original PL intensity after storage under ambient conditions for 30 days. Even after 5000 min of UV-light irradiation, 34.2% of the initial PL intensity was retained.2.7 Double perovskites
Recently, it was reported that Pb2+ cation could be substituted with one monovalent M+ and one trivalent M′3+ cation to form 3D double perovskites AMM′X6 (A = Cs, CH2NH3; M = Ag; M′ = Bi, Sb; X = Cl, Br, I) (Fig. 7a).95–97 Cs2AgBiBr6 with indirect bandgap of 1.95 eV was reported by Slavney et al.98 The lifetime of the intermediate progress in powders is much shorter than that in single crystals due to the higher number of defects and surface states in the powder, indicating that the short and intermediate progresses originate from the trap and surface state emissions, respectively. The radiative recombination lifetime of Cs2AgBiBr6 was higher than that of MAPbI3 films. Further, Cs2AgBiBr6 exhibited better stability against light and moisture than MAPbI3.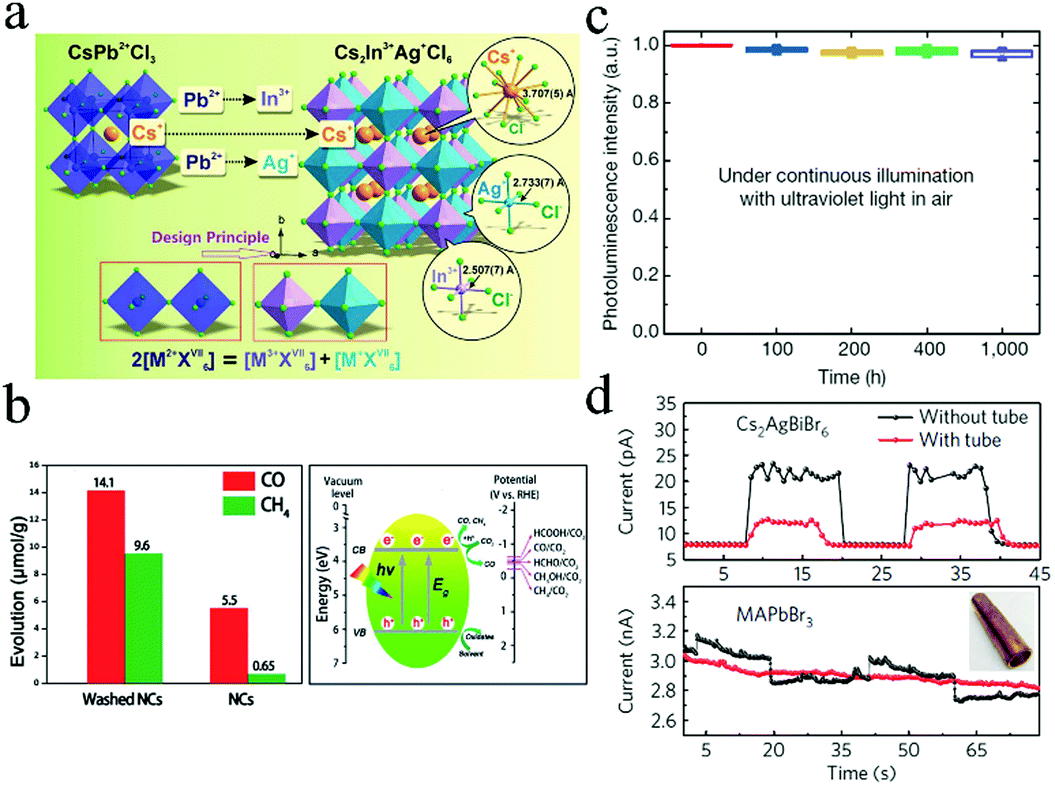 | ||
| Fig. 7 (a) Schematic illustration of Pb substituted by M3+ + M+. Reprinted with permission from ref. 97. Copyright 2017 The Royal Society of Chemistry. (b) Photocatalytic CO2 reduction performance of Cs2AgBiBr6. Reprinted with permission from ref. 100. Copyright 2018 John Wiley and Sons. (c) Stability of Cs2AgInCl6 under continuous illumination. Reprinted with permission from ref. 102. Copyright 2018 Springer Nature. (d) Responses of CsAgBiBr6 and MAPbBr3 with and without tubes, respectively. Reprinted with permission from ref. 43. Copyright 2017 Springer Nature. | ||
McClure et al. synthesized Cs2AgBiBr6 and Cs2AgBiCl6 from both solid state and solution routes.99 The presence of Ag reduced the bandgap and resulted in indirect bandgap. The bandgaps of 2.19 eV and 2.77 eV were determined for Cs2AgBiBr6 and Cs2AgBiCl6, respectively. The reflectance spectrum and XRD patterns of Cs2AgBiCl6 changed marginally when the samples were exposed to both light and air for 4 weeks. Zhou et al. synthesized Cs2AgBiBr6 NCs with a cubic shape and high crystallinity via a hot-injection method.100 These NCs exhibited stability against 55% relative humidity (RH) for 90 days and retained their structure in low-polarity solutions up to 3 weeks. These Cs2AgBiBr6 NCs were used in the photochemical conversion of CO2, achieving total electron consumption of 105 μmol g−1 under AM 1.5G illumination for 6 h (Fig. 7b). Zhou et al. synthesized pure Cs2AgInCl6 crystals with dimensions from several micrometers to several millimeters by a hydrothermal method.97 The UV-vis measurement results indicated that the direct bandgap of Cs2AgInCl6 crystals was 3.23 eV, which was in good agreement with the result obtained from the band structure and optical absorption calculations (3.33 eV). Furthermore, the crystals exhibited no obvious changes when stored under both light and moisture conditions for 26 days. The double perovskite was relatively stable at about 400 °C.
Cs2AgSbCl6 and Cs2AgSbCl6/TiO2 heterostructure nanoparticles have been fabricated to investigate the interfacial effect.103 The Cs4Cl4–TiO2 interface enhanced the separation of photoinduced carriers, which decreased the bandgap and improved light absorption. Therefore, light absorption exhibited a significant red-shift. Moreover, the interfacial states facilitated the tunneling of the photoinduced electrons from the double perovskite layer to TiO2 to improve the PCE of devices. Gao et al. prepared smooth and highly crystalline Cs2AgBiBr6 film when isopropanol (IPA) was used as the antisolvent.101 The crystallinity increased with increased temperatures up to 300 °C. The fabricated devices based on Cs2AgBiBr6 films achieved PCE of 2.23% with Voc = 1.01 V, Jsc = 3.19 mA cm−2, and FF = 0.69. In addition, the solar cells exhibited long-term stability when stored in air for over 10 days. Tang and coworkers reported double-perovskite Cs2AgInCl6 with efficient and stable white-light emissions.102 They revealed that the PL efficiency of Cs2AgInCl6 could be improved by three orders of magnitude by alloying sodium cations. In addition, Cs2(Ag0.60Na0.40)InCl6 achieved 86 ± 5% quantum efficiency and worked for over 1000 h (Fig. 7c). Cs2AgBiBr6 single crystals were applied in X-ray detectors with low detection limits.43 Such a detector exhibited a low detection limit of 59.7 nGyair s−1, which was comparable to that of detectors based on MAPbBr3 single crystals (0.036 μGyair s−1). Furthermore, as shown in Fig. 7d, devices based on Cs2AgBiBr6 exhibited strong response after passing through a thick copper tube, while MAPbBr3 control devices showed no response under the same conditions, indicating the potential industrial applications of Cs2AgBiBr6-based detectors.
3. Conclusions and outlook
In summary, although hybrid lead halide perovskite materials have made considerable progress in optoelectronic applications, there are still several challenges for commercial applications. The intrinsic toxicity of lead perovskites and instability of the organic components can be overcome by all-inorganic lead-free perovskites. Here, we summarize the recent progresses made in all-inorganic lead-free perovskites that can be used for optoelectronic applications. Several research efforts have been devoted toward substituting lead with different elements. Sn-Based perovskites exhibited narrower direct bandgap as compared to their Pb analogues, but the intrinsic instability of Sn2+ induced a serious self-doping problem, which significantly affected the performance of Sn-based perovskites. Bi- and other transition-metal-based lead-free perovskites showed excellent chemical stability, but there are still many challenges. Bi3+- and Sb3+-based perovskites usually exhibit low-dimension crystal phases, which account for their enhanced stabilities, but this also leads to wider bandgaps and lower PV performances. The main problem in novel double perovskites with Bi and Ag are their indirect bandgap, which is undesirable in photovoltaic applications, but they exhibited promising potential for other optoelectronic applications, particularly white-light PL. Currently, all-inorganic lead-free perovskite-based solar cells have exhibited far lower PV performances as compared to hybrid and all-inorganic lead halide perovskites (Table 2). Therefore, much more experimental and theoretical research efforts need to be devoted toward understanding the electrical and optical properties of such lead-free perovskites. Further, new charge transport materials as well as synthesis technologies need to be developed to match the energy level of lead-free perovskites to improve their photovoltaic performance.| Absorber | PCE (%) | V oc (V) | J sc (mA cm−2) | FF (%) | Ref. |
|---|---|---|---|---|---|
| CsSnI3 | 4.81 | 0.38 | 25.71 | 49 | 65 |
| CsSnI3 | 2.76 | 0.43 | 12.3 | 39 | 61 |
| CsSnI3 | 3.31 | 0.52 | 10.21 | 62 | 68 |
| CsSnI3 | 3.26 | 0.40 | 11.25 | 56 | 59 |
| CsSnI2.9Br0.1 | 1.76 | 0.22 | 24.16 | 33 | 58 |
| CsSnI3 | 2.02 | 0.24 | 22.70 | 37 | 57 |
| CsSnBr3 | 3.04 | 0.36 | 13.96 | 59 | 54 |
| CsSnBr3 | 2.1 | 0.41 | 9 | 58 | 60 |
| CsSnBr3 | 0.55 | 0.45 | 2.4 | 55 | 62 |
| CsSnIBr2 | 3.0 | 0.33 | 16.7 | 53 | 63 |
| CsSnCl3 | 9.66 | 0.87 | 19.82 | 56 | 70 |
| CsSnBr3 | 10.46 | 0.85 | 21.23 | 58 | 70 |
| CsSnI3 | 12.96 | 0.86 | 23.2 | 65 | 70 |
| Cs2SnI6 | 1.47 | 0.36 | 6.75 | 59 | 73 |
| Cs2SnI5Br1 | 1.60 | 0.44 | 6.57 | 55 | 73 |
| Cs2SnI4Br2 | 2.02 | 0.56 | 6.22 | 57 | 73 |
| Cs2SnI2Br4 | 1.08 | 0.57 | 3.41 | 54 | 73 |
| Cs2SnI1Br5 | 0.002 | 0.57 | 0.01 | 37 | 73 |
| CsGeI3 | 4.94 | 0.51 | 18.78 | 51 | 77 |
| CsGeBr3 | 4.92 | 0.48 | 19.49 | 52 | 77 |
| CsGeCl3 | 2.57 | 0.35 | 18.57 | 40 | 77 |
| Cs3Bi2I9 | 1.09 | 0.85 | 2.15 | 60 | 79 |
| CsBi3I10 | 0.40 | 0.31 | 3.40 | 38 | 81 |
| Cs2TiBr6 | 3.28 | 1.02 | 5.69 | 56 | 91 |
| Cs2AgBiBr6 | 2.23 | 1.01 | 3.19 | 69 | 101 |
Although the photovoltaic performance of all-inorganic lead-free perovskites still needs to go a long way to be able to compete with that of hybrid lead halide perovskites, such all-inorganic lead-free perovskites are still promising for use in other optoelectronic applications. Therefore, it still requires additional research efforts including both theoretical predictions and experimental optimizations to develop all-inorganic lead-free perovskites with high performance and good stability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (Grant 51861145101, 21777096), Huoyingdong Grant (151046), Shanghai Shuguang Grant (17SG11).References
- A. Miyata, A. Mitioglu, P. Plochocka, O. Portugall, J. T.-W. Wang, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Nat. Phys., 2015, 11, 582 Search PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- J. Huang, Y. Yuan, Y. Shao and Y. Yan, Nat. Rev. Mater., 2017, 2, 17042 CrossRef CAS.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- B. Jeong, H. Han, J. Choi Yung, H. Cho Sung, H. Kim Eui, W. Lee Seung, S. Kim Jong, C. Park, D. Kim and C. Park, Adv. Funct. Mater., 2018, 28, 1706401 CrossRef.
- K. Chen, X. Deng, G. Dodekatos and H. Tüysüz, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS PubMed.
- H. Wang and D. H. Kim, Chem. Soc. Rev., 2017, 46, 5204–5236 RSC.
- S. F. Leung, K. T. Ho, P. K. Kung, V. K. S. Hsiao, H. N. Alshareef, Z. L. Wang and J. H. He, Adv. Mater., 2018, 30, 1704611 CrossRef PubMed.
- W. Zhang, G. E. Eperon and H. J. Snaith, Nat. Energy, 2016, 1, 16048 CrossRef CAS.
- F. P. García de Arquer, A. Armin, P. Meredith and E. H. Sargent, Nat. Rev. Mater., 2017, 2, 16100 CrossRef.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.8b00336.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Gratzel and N.-G. Park, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- H. Chen, F. Ye, W. Tang, J. He, M. Yin, Y. Wang, F. Xie, E. Bi, X. Yang, M. Gratzel and L. Han, Nature, 2017, 550, 92–95 CAS.
- W. S. Yang, B. W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2016, 2, 16177 CrossRef.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- N.-G. Park, M. Grätzel, T. Miyasaka, K. Zhu and K. Emery, Nat. Energy, 2016, 1, 16152 CrossRef CAS.
- W. J. Yin, T. Shi and Y. Yan, Adv. Mater., 2014, 26, 4653–4658 CrossRef CAS PubMed.
- X.-G. Zhao, J.-H. Yang, Y. Fu, D. Yang, Q. Xu, L. Yu, S.-H. Wei and L. Zhang, J. Am. Chem. Soc., 2017, 139, 2630–2638 CrossRef CAS PubMed.
- R. E. Brandt, V. Stevanović, D. S. Ginley and T. Buonassisi, MRS Commun., 2015, 5, 265–275 CrossRef CAS.
- P. V. Kamat, J. Bisquert and J. Buriak, ACS Energy Lett., 2017, 2, 904–905 CrossRef CAS.
- Z. Shi, J. Guo, Y. Chen, Q. Li, Y. Pan, H. Zhang, Y. Xia and W. Huang, Adv. Mater., 2017, 29, 1605005 CrossRef PubMed.
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. H. Chang and M. G. Kanatzidis, Nat. Photonics, 2014, 8, 489–494 CrossRef CAS.
- N. K. Noel, S. D. Stranks, A. Abate, C. Wehrenfennig, S. Guarnera, A.-A. Haghighirad, A. Sadhanala, G. E. Eperon, S. K. Pathak, M. B. Johnston, A. Petrozza, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 3061–3068 RSC.
- R. L. Z. Hoye, R. E. Brandt, A. Osherov, V. Stevanović, S. D. Stranks, M. W. B. Wilson, H. Kim, A. J. Akey, J. D. Perkins, R. C. Kurchin, J. R. Poindexter, E. N. Wang, M. G. Bawendi, V. Bulović and T. Buonassisi, Chem. – Eur. J., 2015, 22, 2605–2610 CrossRef PubMed.
- M. Leng, Y. Yang, K. Zeng, Z. Chen, Z. Tan, S. Li, J. Li, B. Xu, D. Li, M. P. Hautzinger, Y. Fu, T. Zhai, L. Xu, G. Niu, S. Jin and J. Tang, Adv. Funct. Mater., 2018, 28, 1704446 CrossRef.
- M. Leng, Y. Yang, Z. Chen, W. Gao, J. Zhang, G. Niu, D. Li, H. Song, J. Zhang, S. Jin and J. Tang, Nano Lett., 2018, 18, 6076–6083 CrossRef CAS PubMed.
- T. Leijtens, E. Eperon Giles, K. Noel Nakita, N. Habisreutinger Severin, A. Petrozza and J. Snaith Henry, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- C. Qin, T. Matsushima, T. Fujihara, W. J. Potscavage, Jr. and C. Adachi, Adv. Mater., 2015, 28, 466–471 CrossRef PubMed.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- K. Domanski, E. A. Alharbi, A. Hagfeldt, M. Grätzel and W. Tress, Nat. Energy, 2018, 3, 61–67 CrossRef CAS.
- H. Chen, S. Xiang, W. Li, H. Liu, L. Zhu and S. Yang, Sol. RRL, 2018, 2, 1700188 CrossRef.
- G. E. Eperon, G. M. Paterno, R. J. Sutton, A. Zampetti, A. A. Haghighirad, F. Cacialli and H. J. Snaith, J. Mater. Chem. A, 2015, 3, 19688–19695 RSC.
- J. Huang, M. Lai, J. Lin and P. Yang, Adv. Mater., 2018, 1802856, DOI:10.1002/adma.201802856.
- W. Pan, H. Wu, J. Luo, Z. Deng, C. Ge, C. Chen, X. Jiang, W.-J. Yin, G. Niu, L. Zhu, L. Yin, Y. Zhou, Q. Xie, X. Ke, M. Sui and J. Tang, Nat. Photonics, 2017, 11, 726–732 CrossRef CAS.
- P. Gao, M. Grätzel and M. K. Nazeeruddin, Energy Environ. Sci., 2014, 7, 2448–2463 RSC.
- S. Chatterjee and A. J. Pal, J. Mater. Chem. A, 2018, 6, 3793–3823 RSC.
- A. Babayigit, A. Ethirajan, M. Muller and B. Conings, Nat. Mater., 2016, 15, 247–251 CrossRef CAS PubMed.
- D. Yang, J. Lv, X. Zhao, Q. Xu, Y. Fu, Y. Zhan, A. Zunger and L. Zhang, Chem. Mater., 2017, 29, 524–538 CrossRef CAS.
- X. G. Zhao, J. H. Yang, Y. Fu, D. Yang, Q. Xu, L. Yu, S. H. Wei and L. Zhang, J. Am. Chem. Soc., 2017, 139, 2630–2638 CrossRef CAS PubMed.
- I. Chung, J.-H. Song, J. Im, J. Androulakis, C. D. Malliakas, H. Li, A. J. Freeman, J. T. Kenney and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 8579–8587 CrossRef CAS PubMed.
- L.-Y. Huang and W. R. L. Lambrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 165203 CrossRef.
- L. Ma, F. Hao, C. C. Stoumpos, B. T. Phelan, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 14750–14755 CrossRef CAS PubMed.
- J. Liu, M. Ozaki, S. Yakumaru, T. Handa, R. Nishikubo, Y. Kanemitsu, A. Saeki, Y. Murata, R. Murdey and A. Wakamiya, Angew. Chem., Int. Ed., 2018, 57, 13221–13225 CrossRef CAS PubMed.
- L. Qian, Y. Sun, M. Wu, C. Li, D. Xie, L. Ding and G. Shi, Nanoscale, 2018, 10, 6837–6843 RSC.
- T. B. Song, T. Yokoyama, C. C. Stoumpos, J. Logsdon, D. H. Cao, M. R. Wasielewski, S. Aramaki and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 836–842 CrossRef CAS PubMed.
- I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, Nature, 2012, 485, 486 CrossRef CAS PubMed.
- P. Xu, S. Chen, H.-J. Xiang, X.-G. Gong and S.-H. Wei, Chem. Mater., 2014, 26, 6068–6072 CrossRef CAS.
- M. H. Kumar, S. Dharani, W. L. Leong, P. P. Boix, R. R. Prabhakar, T. Baikie, C. Shi, H. Ding, R. Ramesh, M. Asta, M. Graetzel, S. G. Mhaisalkar and N. Mathews, Adv. Mater., 2014, 26, 7122–7127 CrossRef CAS PubMed.
- D. Sabba, H. K. Mulmudi, R. R. Prabhakar, T. Krishnamoorthy, T. Baikie, P. P. Boix, S. Mhaisalkar and N. Mathews, J. Phys. Chem. C, 2015, 119, 1763–1767 CrossRef CAS.
- K. P. Marshall, M. Walker, R. I. Walton and R. A. Hatton, Nat. Energy, 2016, 1, 16178 CrossRef CAS.
- S. Gupta, T. Bendikov, G. Hodes and D. Cahen, ACS Energy Lett., 2016, 1, 1028–1033 CrossRef CAS.
- K. P. Marshall, R. I. Walton and R. A. Hatton, J. Mater. Chem. A, 2015, 3, 11631–11640 RSC.
- D. Moghe, L. Wang, C. J. Traverse, A. Redoute, M. Sponseller, P. R. Brown, V. Bulović and R. R. Lunt, Nano Energy, 2016, 28, 469–474 CrossRef CAS.
- W. Li, J. Li, J. Li, J. Fan, Y. Mai and L. Wang, J. Mater. Chem. A, 2016, 4, 17104–17110 RSC.
- F. Zuo, S. T. Williams, P.-W. Liang, C.-C. Chueh, C.-Y. Liao and A. K.-Y. Jen, Adv. Mater., 2014, 26, 6454–6460 CrossRef CAS PubMed.
- T.-B. Song, T. Yokoyama, S. Aramaki and M. G. Kanatzidis, ACS Energy Lett., 2017, 2, 897–903 CrossRef CAS.
- A. Wang, X. Yan, M. Zhang, S. Sun, M. Yang, W. Shen, X. Pan, P. Wang and Z. Deng, Chem. Mater., 2016, 28, 8132–8140 CrossRef CAS.
- A. Wang, Y. Guo, F. Muhammad and Z. Deng, Chem. Mater., 2017, 29, 6493–6501 CrossRef CAS.
- N. Wang, Y. Zhou, M.-G. Ju, H. F. Garces, T. Ding, S. Pang, X. C. Zeng, N. P. Padture and X. W. Sun, Adv. Energy Mater., 2016, 6, 1601130 CrossRef.
- T. C. Jellicoe, J. M. Richter, H. F. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Bohm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- L. J. Chen, C. R. Lee, Y. J. Chuang, Z. H. Wu and C. Chen, J. Phys. Chem. Lett., 2016, 7, 5028–5035 CrossRef CAS PubMed.
- B. Lee, C. C. Stoumpos, N. Zhou, F. Hao, C. Malliakas, C.-Y. Yeh, T. J. Marks, M. G. Kanatzidis and R. P. H. Chang, J. Am. Chem. Soc., 2014, 136, 15379–15385 CrossRef CAS PubMed.
- W. Zhu, G. Xin, Y. Wang, X. Min, T. Yao, W. Xu, M. Fang, S. Shi, J. Shi and J. Lian, J. Mater. Chem. A, 2018, 6, 2577–2584 RSC.
- B. Lee, A. Krenselewski, S. I. Baik, D. N. Seidman and R. P. H. Chang, Sustainable Energy Fuels, 2017, 1, 710–724 RSC.
- P. P. Sun, Q. S. Li, L. N. Yang and Z. S. Li, Nanoscale, 2016, 8, 1503–1512 RSC.
- T. Krishnamoorthy, H. Ding, C. Yan, W. L. Leong, T. Baikie, Z. Zhang, M. Sherburne, S. Li, M. Asta, N. Mathews and S. G. Mhaisalkar, J. Mater. Chem. A, 2015, 3, 23829–23832 RSC.
- W. Ming, H. Shi and M.-H. Du, J. Mater. Chem. A, 2016, 4, 13852–13858 RSC.
- L.-J. Chen, RSC Adv., 2018, 8, 18396–18399 RSC.
- O. A. Lozhkina, A. A. Murashkina, V. V. Shilovskikh, Y. V. Kapitonov, V. K. Ryabchuk, A. V. Emeline and T. Miyasaka, J. Phys. Chem. Lett., 2018, 9, 5408–5411 CrossRef CAS PubMed.
- B. W. Park, B. Philippe, X. Zhang, H. Rensmo, G. Boschloo and E. M. Johansson, Adv. Mater., 2015, 27, 6806–6813 CrossRef PubMed.
- B. Yang, J. Chen, F. Hong, X. Mao, K. Zheng, S. Yang, Y. Li, T. Pullerits, W. Deng and K. Han, Angew. Chem., Int. Ed., 2017, 56, 12471–12475 CrossRef CAS PubMed.
- M. B. Johansson, H. Zhu and E. M. Johansson, J. Phys. Chem. Lett., 2016, 7, 3467–3471 CrossRef CAS PubMed.
- Y. Lou, M. Fang, J. Chen and Y. Zhao, Chem. Commun., 2018, 54, 3779–3782 RSC.
- R. D. Nelson, K. Santra, Y. Wang, A. Hadi, J. W. Petrich and M. G. Panthani, Chem. Commun., 2018, 54, 3640–3643 RSC.
- C. Wang, B. Yang, R. Ding, W. Chen, R. Kondrotas, Y. Zhao, S. Lu, Z. Li and J. Tang, APL Mater., 2018, 6, 084801 CrossRef.
- F. Jiang, D. Yang, Y. Jiang, T. Liu, X. Zhao, Y. Ming, B. Luo, F. Qin, J. Fan, H. Han, L. Zhang and Y. Zhou, J. Am. Chem. Soc., 2018, 140, 1019–1027 CrossRef CAS PubMed.
- A. M. Ganose, C. N. Savory and D. O. Scanlon, Chem. Commun., 2016, 53, 20–44 RSC.
- C. Zuo and L. Ding, Angew. Chem., Int. Ed., 2017, 56, 6528–6532 CrossRef CAS PubMed.
- B. Saparov, F. Hong, J.-P. Sun, H.-S. Duan, W. Meng, S. Cameron, I. G. Hill, Y. Yan and D. B. Mitzi, Chem. Mater., 2015, 27, 5622–5632 CrossRef CAS.
- J.-C. Hebig, I. Kühn, J. Flohre and T. Kirchartz, ACS Energy Lett., 2016, 1, 309–314 CrossRef CAS.
- M.-G. Ju, M. Chen, Y. Zhou, H. F. Garces, J. Dai, L. Ma, N. P. Padture and X. C. Zeng, ACS Energy Lett., 2018, 3, 297–304 CrossRef CAS.
- M. Chen, M.-G. Ju, A. D. Carl, Y. Zong, R. L. Grimm, J. Gu, X. C. Zeng, Y. Zhou and N. P. Padture, Joule, 2018, 2, 558–570 CrossRef CAS.
- D. Cortecchia, H. A. Dewi, J. Yin, A. Bruno, S. Chen, T. Baikie, P. P. Boix, M. Gratzel, S. Mhaisalkar, C. Soci and N. Mathews, Inorg. Chem., 2016, 55, 1044–1052 CrossRef CAS PubMed.
- X. Li, X. Zhong, Y. Hu, B. Li, Y. Sheng, Y. Zhang, C. Weng, M. Feng, H. Han and J. Wang, J. Phys. Chem. Lett., 2017, 8, 1804–1809 CrossRef CAS PubMed.
- P. Yang, G. Liu, B. Liu, X. Liu, Y. Lou, J. Chen and Y. Zhao, Chem. Commun., 2018, 54, 11638–11641 RSC.
- G. Volonakis, A. A. Haghighirad, R. L. Milot, W. H. Sio, M. R. Filip, B. Wenger, M. B. Johnston, L. M. Herz, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2017, 8, 772–778 CrossRef CAS PubMed.
- F. X. Wei, Z. Y. Deng, S. J. Sun, F. H. Zhang, D. M. Evans, G. Kieslich, S. Tominaka, M. A. Carpenter, J. Zhang, P. D. Bristowe and A. K. Cheetham, Chem. Mater., 2017, 29, 1089–1094 CrossRef CAS.
- J. Zhou, Z. Xia, M. S. Molokeev, X. Zhang, D. Peng and Q. Liu, J. Mater. Chem. A, 2017, 5, 15031–15037 RSC.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- L. Zhou, Y. F. Xu, B. X. Chen, D. B. Kuang and C. Y. Su, Small, 2018, 14, e1703762 CrossRef PubMed.
- W. Gao, C. Ran, J. Xi, B. Jiao, W. Zhang, M. Wu, X. Hou and Z. Wu, ChemPhysChem, 2018, 19, 1696–1700 CrossRef CAS PubMed.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- W. Deng, Z.-Y. Deng, J. He, M. Wang, Z.-X. Chen, S.-H. Wei and H.-J. Feng, Appl. Phys. Lett., 2017, 111, 151602 CrossRef.
| This journal is © the Partner Organisations 2019 |