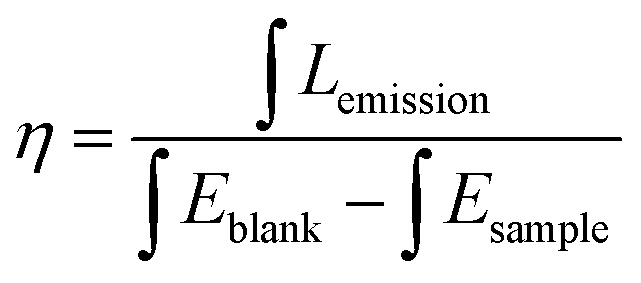DOI:
10.1039/C8QM00586A
(Research Article)
Mater. Chem. Front., 2019,
3, 579-586
Lanthanide-centered luminescence evolution and potential anti-counterfeiting application of Tb3+/Eu3+ grafted melamine cyanurate hydrogen-bonded triazine frameworks†
Received
15th November 2018
, Accepted 16th December 2018
First published on 18th December 2018
Abstract
A novel type of Tb3+/Eu3+ grafted melamine cyanurate hydrogen-bonded triazine framework phosphor (MCA:Ln) has been successfully obtained through an improved co-precipitation route. The actual percentage of Ln3+ ions is less than 1%. The obtained MCA:Ln phosphors show high thermo-stability (330 °C under air) and bright lanthanide (Ln)-centered photoluminescence. The Ln-centered luminescence evolution of MCA:Ln was systematically investigated by steady state excitation and emission spectra, fluorescence decay measurements, TG, XRD, and FT-Raman technology. Moreover, polyvinyl alcohol (PVA)–MCA:Ln mixtures demonstrated the potential to be used as colorful emission inks in the field of anti-counterfeiting.
1. Introduction
The design of a new type of phosphor from organic materials could open the way to a new generation of organic display devices, triboluminescent materials, and thermosensors.1,2 Supramolecular organic frameworks based on weak molecular non-covalent forces such as hydrogen-bonds, π–π interactions, van der Waals interactions, or electrostatic interactions have attracted significant attention.3,4 The extensive hydrogen-bonded frameworks and strong intralayer π–π interactions enable special optical properties of hydrogen-bonded organic frameworks (HOFs), such as the ultralong organic phosphorescence, and the broad solid-state excitation and emission bands.5–7 Melamine and cyanuric acid can form a crystalline melamine cyanurate (MCA) supramolecular framework, interconnected similarly to the hydrogen bonds between guanine and cytosine in DNA.8 Compared with other HOFs, the difference is that the basic building block of MCA is the triazine unit (C3N3). MCA is always used as a precursor for the synthesis of graphitic carbon nitride (g-C3N4) via low temperature thermal condensation between 430![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C and 650°C.9 g-C3N4 and its functional derivatives, as fascinating metal-free semiconductors, have been widely proposed for multifarious photochemical and electrochemical applications, aiming at an affordable clean energy future.10,11 As shown in Scheme 1, a MCA triazine framework possesses rosette-like cavities of approximately 0.4 nm diameter, which are large enough to host small molecules and ions.12 Several papers have studied the possibility of tuning the catalytic activities of MCA by doping Cu2+, Er3+, Tm3+, Pd, and Au.13–15 However, there are few reports about the luminescence properties of MCA triazine frameworks, except that reported by Gao et al., who synthesized blue-emitting MCA complexes by a simple two-step procedure.16
°C and 650°C.9 g-C3N4 and its functional derivatives, as fascinating metal-free semiconductors, have been widely proposed for multifarious photochemical and electrochemical applications, aiming at an affordable clean energy future.10,11 As shown in Scheme 1, a MCA triazine framework possesses rosette-like cavities of approximately 0.4 nm diameter, which are large enough to host small molecules and ions.12 Several papers have studied the possibility of tuning the catalytic activities of MCA by doping Cu2+, Er3+, Tm3+, Pd, and Au.13–15 However, there are few reports about the luminescence properties of MCA triazine frameworks, except that reported by Gao et al., who synthesized blue-emitting MCA complexes by a simple two-step procedure.16
 |
| | Scheme 1 Chemical structure of a MCA hydrogen bonded triazine framework. | |
On the other hand, trivalent lanthanide ions (Ln3+) have very narrow emission lines, showing large Stokes shifts, thus preventing the reabsorption of the emitted light.17,18 This has led to Ln3+ being used in a large variety of applications related to light emission, such as fluorescent lighting, lasers, and so on.19 However the luminescence of Ln3+ ions is a result of transitions within the partially filled 4f shells.20 As these transitions are parity-forbidden this leads to low molar absorption coefficients of Ln3+ ions. Many Ln3+ doped polyoxometalates (POMs), Ln3+ doped metal–organic frameworks (MOFs), Ln3+ doped inorganic materials, and Ln coordination polymers have been created in order to overcome the low molar absorption coefficients and allow excitation of the Ln3+ ions through charge-transfer bands or organic ligands.21–23 Yet among the various studied compounds, Ln MOFs or Ln POMs doped with only a low percentage (less than 1%) of emissive Ln ions are not common.24,25 And the application of luminescent Ln complexes in photonic applications requires thermostable structures, such as material molding processes and solder dissolution processes for the construction of electronic devices.26,27 In general, the degradation of the organic components of typical luminescent organic molecules or Ln MOFs usually starts at moderate temperatures (around 300 °C) under air, so there have been only limited MOFs which were reported to be stable above 300 °C, and nearly none of them exhibit good luminescent behaviors at high temperature.28,29 The MCA triazine framework has excellent thermal stability, showing an endothermic decomposition above 350°C.30 Only few works concerning the combination between the hydrogen-bonded triazine framework and Ln3+ ions have been reported to date.31,32 The photoluminescence properties of a Ln3+ incorporated hydrogen-bonded triazine framework (HTF:Ln) are determined not only by the Ln3+ ions but also by the hydrogen bond frameworks and the interlayer π–π interactions of triazines.33 To the best of our knowledge, there are no reports about the structure and Ln-centered emission evolution of HTF:Ln. Therefore, it is essential to understand the structure and luminescence properties of HTF:Ln and expand their applications.
Here, we report the synthesis of Tb3+/Eu3+ grafted MCA hydrogen-bonded triazine framework phosphors by improved co-precipitation routes.15 Melamine and cyanuric acid are insoluble in most solvents, so DMSO and chloroform are usually employed as the reaction solvents.9,34 However, heating a water solution above 90 °C significantly improves the solubility of melamine and cyanuric acid, making it possible to carry out the synthesis in water solution. The obtained MCA:Ln phosphors were heat treated at different temperatures (130 °C, 230 °C, 330 °C, and 430 °C) under air. By changing the temperature of heat treatment, bright Ln-centered emissions were observed. Here, the relationship between the material's structure and the variable luminescence of MCA:Ln is systematically investigated by steady state excitation and emission spectra, lifetime measurements, quantum yields, TG, and FT-Raman technology.
2. Results and discussion
2.1. The facile synthesis and structure of the MCA:Ln triazine framework
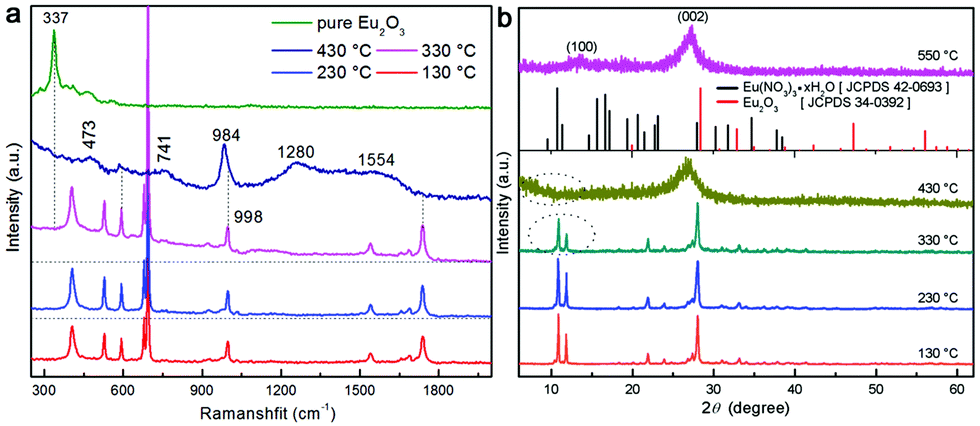
The normalized XRD patterns of pure MCA and MCA:Ln phosphors presented in Fig. 1a show a series of sharp peaks, which correspond to the standard MCA phase (JCPDS: 05-0127). Four well-resolved peaks at lower angles, 10.87, 11.95, 22.15, and 24.09°, are attributed to the in-planar packing between melamine and cyanuric acid through multiple hydrogen bonds, N–H⋯O and N–H⋯N.34 Another strong broad peak at 28.12°, corresponding to a d-spacing of 0.317 nm, supports the π–π stacking of individual MCA nanosheets.9 SEM characterization shown in Fig. S1 (ESI†) reveals that the formed MCA:Ln phosphors have a pillar-like structure with a diameter of a few micrometers and a length of tens to hundreds of micrometers. The TEM images of MCA:Ln shown in Fig. 1 are in good agreement with the SEM analysis. And the higher magnification TEM image inserted in Fig. 1b reveals that the pillar-like particles are composed of three dimensional nanoplates with thickness of 50–70 nm. As in our previous report, the MCA supramolecular framework particles are formed from the lamellar stacking of MCA nanosheets.35
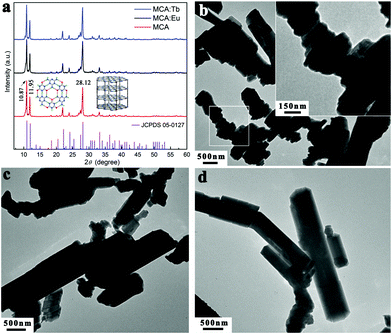 |
| | Fig. 1 (a) Powder XRD patterns of MCA, MCA:Eu, and MCA:Tb [vertical bars at the bottom are the positions of standard MCA (JCPDS 05-0127), inset: schemes showing in-planar packing and π–π stacking of MCA]. (b) TEM image of MCA:Tb [label: showing the stacking of MCA nanoplates], (c) pure MCA, and (d) MCA:Eu. | |
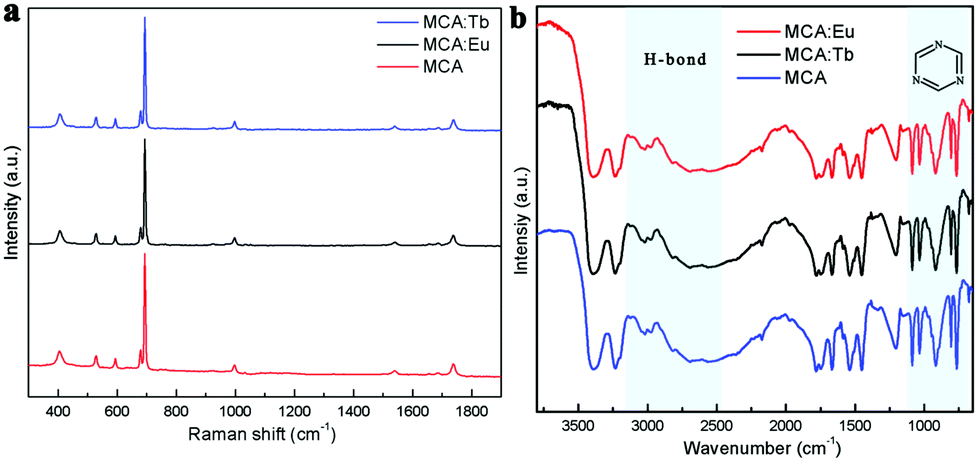
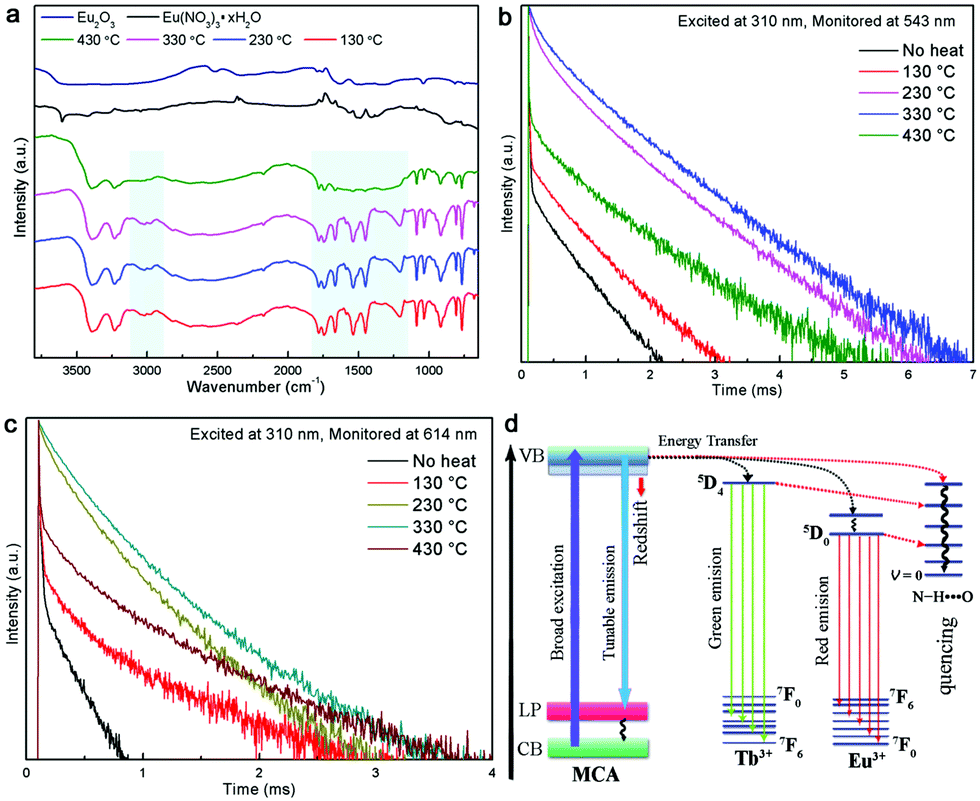
The room temperature (RT) FT-Raman spectra shown in Fig. 2a contain information on the molecular and lattice vibrations of the MCA. Typical Raman peaks at 408, 531, 593, 679, 696, 998, and 1741 cm−1 can be observed, which are in good agreement with the standard Raman spectrum of MCA.36 The most intense double peaks at 679 and 696 cm−1 are related to the vibration of the triazine ring, more specifically they are assigned to the in-plane deformation vibration of the ring and the out-of-plane bending vibration of the ring, which is in accordance with the in-planar packing and lamellar stacking structure between melamine and cyanuric acid.37,38 Four weak peaks at 408, 531, 593, and 998 cm−1 are attributed to the quadrant out-of-plane vibration of the ring, the side-chain in-plane C–N bending, the bending vibration of the ring, and the in-plane radial vibration of the triazine ring, respectively.39 Another weak peak at 1741 cm−1 originates from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration. The RT diffuse reflectance infrared Fourier transform spectra (DRIFTS) shown in Fig. 2b give infornation on the chemical groups of the MCA:Ln phosphors. The broad band around 2600 cm−1 and medium peak at 3041 cm−1 correspond to the amide N–H group interacting via hydrogen bonding with oxygen atoms within cyanuric acid.40 Several sharp peaks in the region 1000–650 cm−1 can be assigned to the triazine ring vibration modes. As is reported, there is no obvious difference in the FT-Raman or intermediate infrared spectra of the metal ion doped triazine or heptazine framework.13,14,35,41
O stretching vibration. The RT diffuse reflectance infrared Fourier transform spectra (DRIFTS) shown in Fig. 2b give infornation on the chemical groups of the MCA:Ln phosphors. The broad band around 2600 cm−1 and medium peak at 3041 cm−1 correspond to the amide N–H group interacting via hydrogen bonding with oxygen atoms within cyanuric acid.40 Several sharp peaks in the region 1000–650 cm−1 can be assigned to the triazine ring vibration modes. As is reported, there is no obvious difference in the FT-Raman or intermediate infrared spectra of the metal ion doped triazine or heptazine framework.13,14,35,41
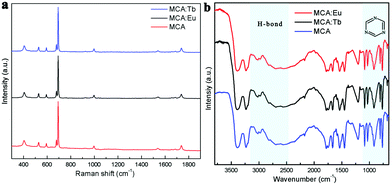 |
| | Fig. 2 (a) FT-Raman spectra of MCA, MCA:Tb, and MCA:Eu. (b) DRIFTS spectra of MCA, MCA:Tb, and MCA:Eu. | |
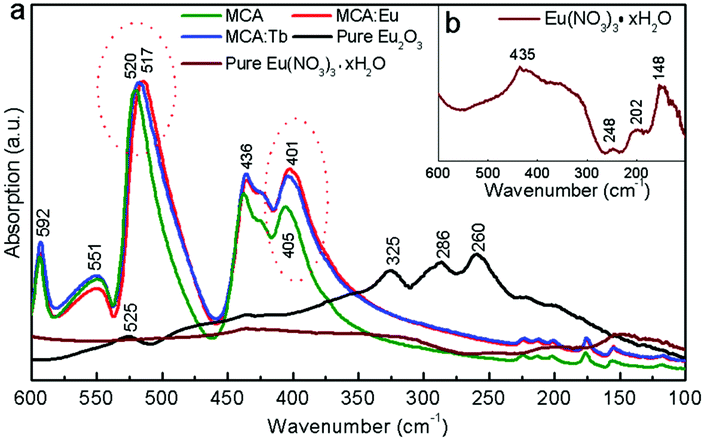
In order to investigate the possible position of Ln3+ ions in the MCA framework, MCA:Ln samples were also prepared through a centrifugal route (Scheme S1, ESI†). The obtained samples are labelled MCA:Eu-C and MCA:Tb-C. As shown in Fig. S2 (ESI†) no characteristic peaks of Eu2O3, Eu(NO3)3·xH2O, melamine, or cyanuric acid were found in the XRD patterns. The ICP-MS analysis was carried out to compare the concentrations of Ln3+ ions in the samples synthesised by two routes. As shown in Tables S1 (ESI†), only 0.111% Eu3+ or 0.121% Tb3+ ions were doped into the MCA framework. It is therefore clear that most of the Ln3+ ions grafted onto the surface of MCA and could be washed away in this synthetic route. Despite the low doping percentage of the Ln3+ ions, the characteristic emisison of Eu3+ and Tb3+ could be observed in the MCA:Eu-C and MCA:Tb-C samples, respectively (Fig. S3, ESI†). So the potential structure of MCA:Ln is that the Ln3+ ions are grafted with the MCA triazine framework by weak chemical bonds. It is known that the far infrared measurements in the 200–700 cm−1 region are a convincing technology to study the coordination structure or other interactions of Ln 3+ ions in the Ln doped complexes. The vacuum far infrared spectra of pure MCA, MCA:Ln, pure Eu2O3, Eu(NO3)3·xH2O were collected and are presented in Fig. 3. In the far IR spectrum of MCA:Ln, the absorption bands ranging from 100–600 cm−1 are similar to those of the as-synthesized pure MCA. Furthermore, the strong Eu–O interactions of pure Eu2O3 observed at 525, 325, 286, and 260 cm−1 and pure Eu(NO3)3·xH2O (Fig. 3b) observed at 435, 248, 202, and 148 cm−1 are not found in the spectra of MCA:Eu. Our results show that the side-chain in-plane bend vibration of C–NHx is shifted to a lower wavenumber from 520 to 517 cm−1, and the quadrant out-of-plane vibration of the triazine ring also is shifted to a lower wavenumber from 405 to 401 cm−1.39 Based on these measurements we believe that the Eu3+ ions in the samples are most probably grafted onto the MCA triazine framework by C–N–EuIII interactions.42
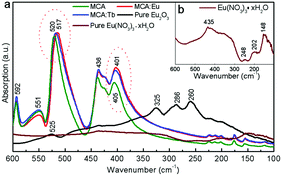 |
| | Fig. 3 (a) The vacuum far infrared spectra of MCA, MCA:Eu, MCA:Tb, Eu2O3, and Eu(NO3)3·xH2O, inset: (b) the enlarged spectrum of Eu(NO3)3·xH2O. | |
2.2. The lanthanide-centered luminescence evolution of the MCA:Ln triazine framework
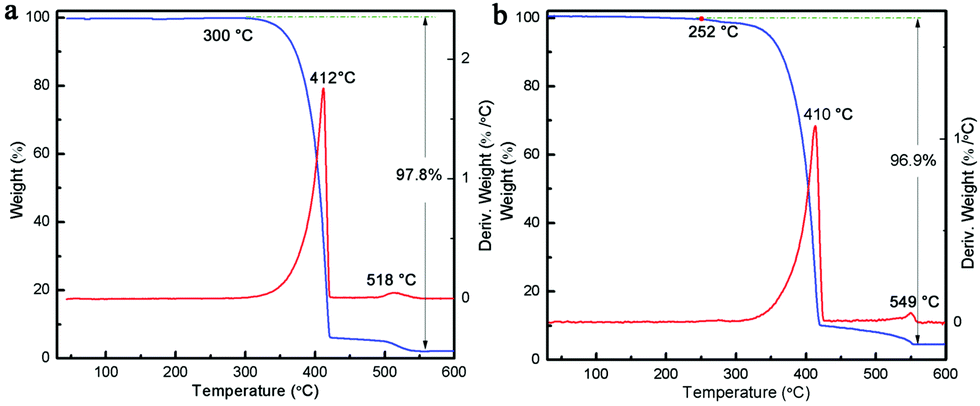
It is known that upon heating, MCA undergoes condensation reactions with the detachment of ammonia and gradual linking of cyanuric rings through imide bridges.40 Therefore, a thermal analysis of pure MCA and the as-synthesized MCA:Ln phosphors was carried out in order to investigate the structure evolutions in physical and chemical state in relation to the increasing temperature. Fig. 4a shows the TG and DTG curves recorded for pure MCA. The TG curve of MCA exhibits mass losses in two stages, which indicates that decomposition of MCA takes place gradually. The large step starting from about 300 °C to 418 °C corresponded to the liberation of ammonia and carbon dioxide release, and the decomposition of MCA to g-C3N4.9 The second step beginning after 540 °C was attributed to the decomposition of g-C3N4. Accordingly, the DTG curve of pure MCA shows two distinct peaks at 412 and 518 °C in the same regions and their relative intensities. Obviously the TG and DTG curves recorded for the synthesized MCA:Tb (Fig. 4b) and MCA:Eu (Fig. S4, ESI†) have similar profiles to the pure MCA. However the introduction of europium nitrate or terbium nitrate leads to the initial weight loss of MCA:Ln at a lower temperature (about 250 °C). The total weight losses of MCA, MCA:Tb, and MCA:Eu are about 97.8%, 96.9%, and 96.2%, respectively, which is in agreement with the ICP-MS analysis.
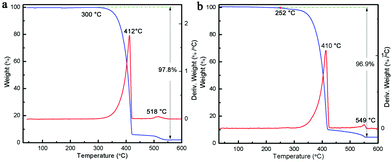 |
| | Fig. 4 TG and DTG curves of (a) pure MCA and (b) MCA:Tb. | |
In order to investigate the relationship between the Ln-centered luminescence and the thermal stability of MCA:Ln, the MCA:Ln phosphors were heated at different temperatures in air. Fig. 5 presents the PL excitation and emission spectra of MCA:Eu and MCA:Tb without heating and after heating from 130 °C to 430 °C. The enlarged PL spectra of MCA:Ln without heating and after heating at 130 °C are aslo inserted in Fig. 5. All labelled peaks have been assigned to the appropriate transitions in Table S2 (ESI†). In the excitation spectrum of the MCA:Eu sample without heating, a series of very weak sharp peaks are observed, which can be assigned to the characteristic f–f transitions of Eu3+. The spectrum of the 130 °C product shows similar sharp peaks as well as a weak broad band in the 300–380 nm region resulting from the charge-transfer (CT) transitions (4fn → 4fn+1 L−1, where L = ligand).43 When the processing temperature increases to 230 °C and 330 °C, most of the weak sharp peaks disappear and a strong broad band between 260 and 420 nm with a maximum at around 331 nm appears. As is reported, the broad strong band in the case of the europium grafted MCA triazine framework can be ascribed to originate from Eu–N CT transitions, due to the fact that the CT band of Eu–O always appears around 250–300 nm (40000 cm−1).43 It is also in good agreement with the structure analysis of MCA:Eu that the Eu3+ ions form the C–N–EuIII interaction with the MCA triazine framework. The strong charge transfer band in the excitation spectra indicates that the energy transfer from the MCA matrix to Eu3+ ions is more efficient. When the processing temperature increases to 430 °C, the strong broad band between 260 and 420 nm weakens again and the maximum redshifts to 344 nm, which indicate that the high processing temperature damages the CT transition. The emission of the MCA:Eu without heat treatment was observed after exciting at 310 nm due to the Eu–N transition and is shown in Fig. 5b. It could be clearly seen that exciting at 310 nm yielded the weak characteristic emission peaks of Eu and a strong emission band at 355 nm originating from the MCA matrix. In the emission spectrum recorded for the 130 °C product the MCA matrix band is still visible, yet the f–f transition peaks are stronger in intensity. With a temperature increase to 230 °C, the Ln-centered luminescence strongly increases, while the broad emission band originated from the MCA matrix obviously redshifts to 365 nm. For the 330 °C product the strongest characteristic transitions peaks 5D0 → 7F0–4 are present and the MCA matrix band continuously redshifts to 376 nm. When the temperature increases further to 430 °C, the Ln-centered luminescence weakens sharply, and the emission from the MCA matrix obviously increases and shifts towards longer wavelengths at 430 nm.
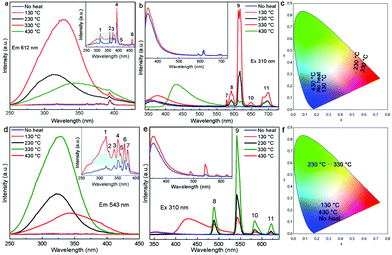 |
| | Fig. 5 PL excition (a) and emission (b) spectra of MCA:Eu after heat treatment at different temperatures. PL excitation (d) and emission (e) spectra of MCA:Tb after heat treatment at different temperatures. The CIE chromaticity diagram of (c) MCA:Eu and (f) MCA:Tb after heat treatment at different temperatures. | |
The PL spectra of MCA:Tb also present similar evolution to the MCA:Eu. As can be seen from Fig. 5d for all the MCA:Tb samples after heating above 130 °C in the excitation spectra only the charge transfer band is present. No f–f transition peaks could be detected in either of the excitation spectra. In the emission spectra (Fig. 5e) recorded after 230 °C and 330 °C heat treatment, respectively the MCA matrix band almost disappears, yet the f–f transition peaks are still very intense. This indicates that the energy transfer from the MCA matrix to Tb3+ ions is more efficient than that of MCA:Eu. The peaks presented in the figures were assigned to the appropriate transitions in Table S3 (ESI†). The CIE chromaticity coordinates of the MCA:Ln phosphors are included in Fig. 5c, f and Table S4 (ESI†). When the heat treatment temperature is lower than 230 °C, the emission colour is in the light blue region. For the products obtained after 230 °C and 330 °C heat treatment, respectively the emission colour shifts to the green or red light region. When the temperature increases further to 430 °C, the emission colour shifts back to blue light region. The absolute overall quantum yield of as-synthesized MCA:Ln after heating at 330 °C was measured by using an integrated sphere with a BENFLEC coating provided by Edinburgh Instruments as follows:44
where
Lemission is the integrated value of the emission spectrum, and
Eblank and
Esample are the integrated values of the “excitation” band of the blank and the excitation band of the sample (since the sample absorbs part of the light, this value will be smaller than
Eblank), respectively. The quantum yield of the as-prepared MCA:Tb after heating at 330 °C is 14.76%, which is higher than that of MCA:Eu after heating at 330 °C (9.52%).
In order to interpretate the Ln-centered emisson mechanism, microstructure evolution of MCA:Ln products after heating under different conditions were investigated by FT-Raman spectra, XRD patterns, and intermediate infrared spectra. As shown in Fig. 6a, the MCA:Eu is still in the MCA form when the processing temperature is lower than 350 °C. It is observed that with increasing temperature the Raman background luminescence is intense. The phenomenon was also observed in the Raman spectra of melamine after thermal condensation at different temperatures.45 As a previous report has shown, it indicates that the crystalline raw material transformed into an amorphous product at high temperature.46 In the Raman spectrum of the 430 °C product several characteristic peaks of g-C3N4 at 473, 741, 984, 1280 and 1554 cm−1 are observed, which indicates that the MCA matrix transforms into the amorphous g-C3N4 phase.47 The typical Raman peak of Eu2O3 at 337 cm−1 (the Fg mode) is not found, which illustrates that europium in the ions forms bond with the carbon nitride matrix. The PXRD patterns of MCA:Tb heated at different temperatures have a similar result to that of FT-Raman. As shown in Fig. 6b, when the heating temperatures remain below 330 °C, the XRD patterns indicate that the MCA:Eu phosphor maintains its MCA phase. For the 330 °C and 430 °C products the typical double peaks of MCA at about 10° obviously fade away and a broad typical peak of g-C3N4 at about 26.8° appears.47 When the temperature increases further to 550 °C, two characteristic peaks of g-C3N4 can be observed. One is the strong peak at 27.2° corresponding to the (002) plane, which is attributed to the interlayer stacking of aromatic segments. The other is the weak peak at 12.8° attributed to the (100) plane, which is related to the in-plane structural packing of carbon nitride units.34 Meanwhile, no characteristic peaks of Eu2O3 and Eu(NO3)3·xH2O are found in the XRD patterns of MCA:Eu. The intermediate infrared spectra of MCA:Tb shown in Fig. 7a also present a similar microstructure evolution. In the infrared spectrum of the 430 °C product several peaks from 1100 to 1700 cm−1 dominate the spectra, corresponding to the characteristic stretching modes of C–N heterocycles of g-C3N4.47 The medium peaks at 3026 and 2977 cm−1 assigned to the NH2 asymmetric stretching vibration are significantly weaker, indicating higher degree of polycondensation which is in accordance with the XRD and FT-Raman results.11 Essentially, the Ln-centered emisson is in parallel with the microstructure evolution of the MCA matrix, and the destruction of the MCA matrix will weaken the Ln-centered emisson.
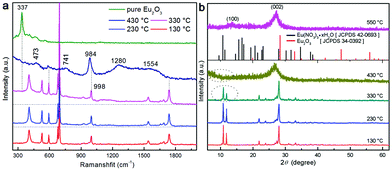 |
| | Fig. 6 (a) FT-Raman spectra of pure Eu2O3 and MCA:Eu after heat treatmet at different temperatures. (b) Normalized PXRD patterns of MCA:Eu MCA:Eu after heat treatment at different temperatures [vertical bars at the bottom are the positions of standard Eu2O3, standard Eu(NO3)3·xH2O]. | |
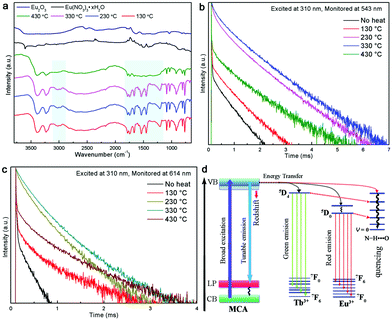 |
| | Fig. 7 (a) Intermediate infrared spectra of pure Eu2O3, Eu(NO3)3·xH2O, and MCA:Eu after heat treatment at different temperatures. (b) The normalized luminescence lifetime curves of MCA:Tb after heating at different temperatures. (c) The normalized luminescence lifetime curves of MCA:Eu after heating at different temperatures. (d) Schematic photoexcited energy transfer evolution of MCA:Ln. | |
The RT luminescence lifetime measurements of the MCA:Ln phosphors were performed and the results are shown in Fig. 7b and c. The experimental data could be fitted well with two exponential decay functions with a fast component τf and a slower component τs. The fitting results are listed in Tables S5 and S6 (ESI†). In general, the slow parameters correspond to the radiative transition rates, while the faster lifetime reflects the nonradiative transition rates, such as the relaxation from excited states to lower energy levels.48 When the processing temperature increases from 130 °C to 230 °C, both the fast and slower components increase in length, but the fast component does so in a more pronounced way: for samples heated at 230 °C it increases about 10 fold compared to the samples heated at 130°. From the previous study, several factors have been associated with the improvement of the lifetime including the optimal doping concentration, the use of inorganic or organic protective shell, or the structural defects and unsaturated bonds.49,50 Obviously, heat treatment at a moderate temperature does not change the doping concentration or add a usefully protective shell for the MCA:Ln material. In general, the naturally hydrated surfaces undergo hydroxyl group desorption during the heat treatment at around 300 °C, which will result in the high density of reactive centres involved in the melamine surface.51 In this scenario, as shown in Fig. 7d, the MCA matrix has a big chance to transfer excited electrons to Ln3+ ions instead of dissipating the energy through non-radiative recombination channels. So the Ln-centered luminescence obviously increases. And the heat treatment temperature of 330 °C redshifts the emission of the MCA host, which also improves the energy transfer from the host to Ln3+ ions, so the fluorescence lifetimes continually increase.52 However when the processing temperature increases to 430 °C, the crystalline MCA transforms into the amorphous g-C3N4. The rapidly decreasing lifetime components probably reflect on the excited energy being reabsorbed by the g-C3N4 matrix instead of passing on the energy to the Ln3+ ion. Recent reports show that the g-C3N4 is not a good matrix for Eu3+ luminescence.41 So the Ln-centered luminescence obviously decreases.
2.3. The potential anti-counterfeiting application of MCA:Ln phosphors
As an illustration of a potential application of these materials, the PVA solution (10 wt%) is used as a “liquid glue” to mix with the MCA and MCA:Ln after being heated at 330 °C. As shown in Fig. 8a, the obtained MCA–PVA mixtures (MCA–PVA) emit bright red, green, and blue luminescence under a UV lamp. The obtained MCA–PVA mixtures could be used directly as an “ink”, to write on weighing paper, and on a banknote. As shown in Fig. 8b–e, the letters ‘‘UGENT’’ written on a weighing paper in MCA–PVA ink show bright colorful luminescence, which suggests that MCA–PVA could be used as a secret information carrier (security ink) on paper documents. To demonstrate the performance of MCA–PVA as an anti-counterfeiting tool, two characters “UT” were handwritten on a banknote in MCA–PVA ink. After drying, the letters are hardly observable under daylight. Upon irradiation with a 302 nm UV lamp, bright green and red emission can be seen.
 |
| | Fig. 8 (a) Photographs of MCA–PVA mixtures under a UV lamp (302 nm). Photographs of letters ‘‘UGENT’’ written on a weighing paper under a 302 nm UV lamp (b)–(d), and a blank paper (e). Handwritten characters ‘‘UT’’ in MCA–PVA ink on a banknote under daylight (f) and UV light (h), and a blank note (g) and (i). | |
3. Experimental
3.1. Materials and measurements
All chemicals were purchased from Sigma-Aldrich or Fisher Scientific and used as received without further purification. Powder X-ray diffraction patterns were measured on a Thermo Scientific ARL X’TRA diffractometer at room temperature. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were recorded on a Thermo Nicolet 6700 spectrometer, equipped with a nitrogen-cooled MCTA detector and a KBr beam splitter at room temperature. The vacuum far infrared spectroscopy measurements were recorded on a Bruker Vertex-80v Fourier-transform infrared spectrometer. Scanning electron microscopy micrographs were obtained on a SEM apparatus, type S-3000N (Hitachi). Transmission electron microscopy (TEM) images were taken on a JEOL-2100 electron microscope. Thermo-gravimetric analysis (TGA) was carried out on a SDT Q600 analyzer with a heating rate of 10 °C min−1. Fourier transform Raman spectroscopy (FT-Raman) measurements were recorded on a Thermo Nicolet 6700 NXR FT-Raman spectrometer with an InGaAs detector at room temperature. The organic elemental analyses were carried out using a thermo-organic elemental analysis flash 2000 apparatus. The luminescence photos of the samples were acquired using a Canon IXUS digital camera, and the samples were placed under a Cole-Parmer laboratory UV lamp, which could operate at either 254, 302, or 365 nm. For the Ln3+ concentrations, 20 mg of MCA based phosphor was digested with 6 mL of 37% HCl (Chem-Lab, Zedelgem) and 2 mL of 65% HNO3 (Chem-Lab, Zedelgem) for 2 h at 100 °C, after which inductively coupled plasma mass spectrometry (ICP-MS) analysis was carried out on an inductively coupled plasma mass spectrometer (Nexion 350, Perkin Elmer, US). The photoluminescence spectra were recorded on an Edinburgh Instruments FLSP 920 UV-vis-NIR spectrofluorimeter with a 450 W xenon lamp as the steady state excitation source. Luminescence decay times were recorded using a 60 W pulsed Xe lamp, operating at a frequency of 100 Hz. All the decay curves could be well-fitted using a double exponential eqn (S1), where I(t) and I0 stand for the luminescence intensity of the MCA based phosphor at time t and time 0, A1 and A2 are constants, and τs and τf correspond to the luminescence lifetimes for the fast and slow components, respectively. After fitting the curves, the average lifetimes (τave) could be calculated using eqn (S2).| | | I(t) = I0 + A1exp(−t/τs) + A2exp(−t/τf) | (S1) |
| | | τave = (A1τs2 + A2τf2)/(A1τs + A2τf) | (S2) |
3.2. Synthesis of MCA based phosphors
All MCA based phosphors were synthesized by a modified previously reported route.15,53 As shown in Scheme S1 (ESI†), to prepare MCA:Tb, 0.086 g (0.25 mmol) Tb(NO3)3·xH2O powder was dissolved in 5 mL of deionized water to obtain a 0.05 mol L−1 Tb(NO3)3 solution. 0.63 g melamine and 0.65 g cyanuric acid were each dissolved in 80 mL of deionized water under stirring at 97 °C, respectively. Then 2 mL of the as-prepared 0.05 mol L−1 Tb(NO3)3 solution was added to the melamine solution under stirring at 97 °C. Next, the cyanuric acid solution was added to the previous mixed solution of melamine and Tb(NO3)3 under stirring at 97 °C. The obtained white mixtures were heated at 97 °C until about 30 mL of the mixture remained. The residues were dried at 80 °C in order to obtain a powder. The obtained white powder was labelled MCA:Tb. In another procedure 0.086 g (0.25 mmol) Tb(NO3)3·xH2O powder was dissolved in 5 mL of deionized water to obtain 0.05 mol L−1 Tb(NO3)3 solution. 0.63 g melamine and 0.65 g cyanuric acid were each dissolved in 80 mL of deionized water under stirring at 97 °C, respectively. Then 2 mL of the as-prepared 0.05 mol L−1 Tb(NO3)3 solution was added to the as-prepared melamine solution under stirring at 97 °C. Next, the as-prepared cyanuric acid solution was added to the previous mixed solution of melamine and Tb(NO3)3 under stirring at 97 °C. The obtained white mixtures were heated at 97 °C until about 30 mL of the mixture remained. It was further centrifuged (3000 rpm) and washed with deionized water three times, and then dried at 80 °C in order to obtain a powder. The obtained white powder was labelled MCA:Tb-C. The Eu3+ doped MCA phosphors and pristine MCA powders were also prepared through similar procedures. The obtained samples were labelled MCA:Eu, MCA:Eu-C, and MCA. A table with the actual Ln3+ concentrations found for each sample (using ICP-MS) is presented in Table S1 (ESI†).
3.3. Thermostability experiments for MCA based phosphors
All MCA:Ln phosphors were tested by a similar route. For example, MCA:Tb in a reinforced glass bottle was placed in the middle region of a furnace chamber. The samples were heated to a designed temperature (130 °C, 230 °C, 330 °C, and 430 °C) at a heating rate of 12 °C min−1 under air, and held for 30 min. After heating, the sample was taken out of the oven and left to cool to room temperature. The obtained samples were labelled MCA:Tb-130, MCA:Tb-230, MCA:Tb-330, and MCA:Tb-430. Other samples were also prepared through a similar procedure.
3.4. Preparation of MCA–PVA emissive inks
2 mL of PVA water solution (10 wt%) was mixed with 0.1 g of MCA-330, MCA:Eu-330, and MCA:Tb-330 powders, respectively. The obtained MCA–PVA mixtures were employed as luminescent inks to write on banknote and weighing paper directly.
4. Conclusions
In summary, in this work novel MCA:Tb3+/Eu3+ hydrogen-bonded triazine frameworks (HOFs) have been successfully synthesized via an improved water based route. These MCA:Ln phosphors possess high thermal stability (330 °C). After heat-treatment at different temperatures, the MCA:Ln phosphors show tunable Ln-centered luminescence due to unfixed energy transfer from the MCA matrix to the Ln3+ ions. Moreover, MCA–PVA mixtures demonstrated the potential to be used as anti-counterfeiting security inks.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
C. Q. Y. acknowledges the China Scholarship Council (CSC, Grant No. 201707565016). A. M. K. and K. L. acknowledge Ghent University's Special Research Fund (BOF) for a Postdoctoral Mandate (Grants No. BOF15/PDO/091, and BOF16/PDO/159, respectively). H. V. thanks the Research Foundation Flanders (FWO, Hercules Foundation, FT-imager project AUGE/13/16) for financial support.
Notes and references
- S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., 2013, 125, 2953–2957 CrossRef.
- M. Allendorf, C. Bauer, R. Bhakta and R. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- P. Li, Y. He, Y. Zhao, L. Weng, H. Wang, R. Krishna, H. Wu, W. Zhou, M. O'Keeffe and Y. Han, Angew. Chem., Int. Ed., 2015, 54, 574–577 CAS.
- J. C. MacDonald and G. M. Whitesides, Chem. Rev., 1994, 94, 2383–2420 CrossRef CAS.
- J.-H. Wang, G.-M. Tang, T.-X. Qin, Y.-T. Wang, Y.-Z. Cui and S. W. Ng, J. Coord. Chem., 2017, 70, 1168–1189 CrossRef CAS.
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling and M. Gu, Angew. Chem., Int. Ed., 2018, 57, 4005–4009 CrossRef CAS PubMed.
- H. Wang, Z. Bao, H. Wu, R.-B. Lin, W. Zhou, T.-L. Hu, B. Li, J. C.-G. Zhao and B. Chen, Chem. Commun., 2017, 53, 11150–11153 RSC.
- W. Xu, M. Dong, H. Gersen, E. Rauls, S. Vázquez-Campos, M. Crego-Calama, D. N. Reinhoudt, I. Stensgaard, E. Laegsgaard and T. R. Linderoth, Small, 2007, 3, 854–858 CrossRef CAS PubMed.
- Y. S. Jun, E. Z. Lee, X. Wang, W. H. Hong, G. D. Stucky and A. Thomas, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- A. Ranganathan, V. Pedireddi and C. Rao, J. Am. Chem. Soc., 1999, 121, 1752–1753 CrossRef CAS.
- F. Jiang, R. Li, J. Cai, W. Xu, A. Cao, D. Chen, X. Zhang, C. Wang and C. Shu, J. Mater. Chem. A, 2015, 3, 19433–19438 RSC.
- R. C. Dante, F. M. Sánchez-Árevalo, P. Chamorro-Posada, J. Vázquez-Cabo, L. Lartundo-Rojas, J. Santoyo-Salazar, R. Mendoza-Cruz, J. J. Velázquez-Salazar, M. J. Arellano-Jiménez and J. E. Samaniego, Fullerenes, Nanotubes, Carbon Nanostruct., 2016, 24, 688–697 CrossRef CAS.
- J. Xu, T. J. Brenner, Z. Chen, D. Neher, M. Antonietti and M. Shalom, ACS Appl. Mater. Interfaces, 2014, 6, 16481–16486 CrossRef CAS PubMed.
- Y. Qiu and L. Gao, Mater. Res. Bull., 2005, 40, 794–799 CrossRef CAS.
- W. Carnall, P. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4424–4442 CrossRef CAS.
- L. Melby, N. Rose, E. Abramson and J. Caris, J. Am. Chem. Soc., 1964, 86, 5117–5125 CrossRef CAS.
- S. F. Mason, R. D. Peacock and B. Stewart, Mol. Phys., 1975, 30, 1829–1841 CrossRef CAS.
- P. Allen, J. Bucher, D. Shuh, N. Edelstein and I. Craig, Inorg. Chem., 2000, 39, 595–601 CrossRef CAS PubMed.
- P. Wang, J.-P. Ma, Y.-B. Dong and R.-Q. Huang, J. Am. Chem. Soc., 2007, 129, 10620–10621 CrossRef CAS PubMed.
- L. Ma, O. R. Evans, B. M. Foxman and W. Lin, Inorg. Chem., 1999, 38, 5837–5840 CrossRef CAS.
- J. Heine and K. Müller-Buschbaum, Chem. Soc. Rev., 2013, 42, 9232–9242 RSC.
- M. Saif, J. Lumin., 2013, 135, 187–195 CrossRef CAS.
- A. M. Kaczmarek, K. Van Hecke and R. Van Deun, Inorg. Chem., 2017, 56, 3190–3200 CrossRef CAS PubMed.
- Y. Hasegawa and T. Nakanishi, RSC Adv., 2015, 5, 338–353 RSC.
- L. N. Puntus, K. J. Schenk and J. C. G. Bünzli, Eur. J. Inorg. Chem., 2005, 4739–4744 CrossRef CAS.
- A. Ramya, D. Sharma, S. Natarajan and M. Reddy, Inorg. Chem., 2012, 51, 8818–8826 CrossRef CAS.
- H. Zhang, L. Zhou, J. Wei, Z. Li, P. Lin and S. Du, J. Mater. Chem., 2012, 22, 21210–21217 RSC.
- H. Huang, K. Zhang, J. Jiang, J. Li and Y. Liu, Polym. Int., 2017, 66, 85–91 CrossRef CAS.
- D. Durham, G. Frost and F. Hart, J. Inorg. Nucl. Chem., 1969, 31, 571–574 CrossRef CAS.
- Y.-F. Zhao, H.-B. Chu, F. Bai, D.-Q. Gao, H.-X. Zhang, Y.-S. Zhou, X.-Y. Wei, M.-N. Shan, H.-Y. Li and Y.-L. Zhao, J. Organomet. Chem., 2012, 716, 167–174 CrossRef CAS.
- H. Zhang, N. Li, C. Tian, T. Liu, F. Du, P. Lin, Z. Li and S. Du, Cryst. Growth Des., 2012, 12, 670–678 CrossRef CAS.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- C. Yang, W. Zhao, X. Yu, H. Liu, J. Liu, R. Van Deun and Z. Liu, Mater. Res. Bull., 2017, 94, 415–422 CrossRef CAS.
- R. Reimschuessel, C. M. Gieseker, R. A. Miller, J. Ward, J. Boehmer, N. Rummel, D. N. Heller, C. Nochetto, G. H. de Alwis and N. Bataller, Am. J. Vet. Res., 2008, 69, 1217–1228 CrossRef CAS PubMed.
- E. Koglin, B. J. Kip and R. J. Meier, J. Chem. Phys., 1996, 100, 5078–5089 CrossRef CAS.
-
L. He, Y. Liu, M. Lin, J. Awika, D. R. Ledoux, H. Li and A. Mustapha, Sensing and Instrumentation for Food Quality and Safety, 2008, vol. 2, pp. 66–71 Search PubMed.
- M. Marchewka, Mater. Lett., 2004, 58, 843–848 CrossRef CAS.
- V. Sangeetha, N. Kanagathara, R. Sumathi, N. Sivakumar and G. Anbalagan, J. Mater., 2013, 2013, 262094 Search PubMed.
- M. Wang, P. Guo, Y. Zhang, C. Lv, T. Liu, T. Chai, Y. Xie, Y. Wang and T. Zhu, J. Hazard. Mater., 2018, 349, 224–233 CrossRef CAS PubMed.
- H. Li, Y. Xia, T. Hu, Q. Deng, N. Du and W. Hou, J. Mater. Chem. A, 2018, 6, 6238–6243 RSC.
- A. Nag, P. J. Schmidt and W. Schnick, Chem. Mater., 2006, 18, 5738–5745 CrossRef CAS.
- H. Zheng, W. Chen, H. Gao, Y. Wang, H. Guo, S. Guo, Z. Tang and J. Zhang, J. Mater. Chem. C, 2017, 5, 10746–10753 RSC.
- Y. Zhang, Q. Pan, G. Chai, M. Liang, G. Dong, Q. Zhang and J. Qiu, Sci. Rep., 2013, 3, 1943 CrossRef PubMed.
- S. Belaya, V. Bakovets, A. Boronin, S. Koshcheev, M. Lobzareva, I. Korolkov and P. Stabnikov, Inorg. Mater., 2014, 50, 379–386 CrossRef CAS.
- I. Papailias, T. Giannakopoulou, N. Todorova, D. Demotikali, T. Vaimakis and C. Trapalis, Appl. Surf. Sci., 2015, 358, 278–286 CrossRef CAS.
- Z. Gan, Y. Shan, J. Chen, Q. Gui, Q. Zhang, S. Nie and X. Wu, Nano Res., 2016, 9, 1801–1812 CrossRef CAS.
- P. Ricci, M. Salis, R. Corpino, C. Carbonaro, E. Fortin and A. Anedda, J. Appl. Phys., 2010, 108, 043512 CrossRef.
- L. Stagi, J. A. De Toro, A. Ardu, C. Cannas, A. Casu, S. S. Lee and P. C. Ricci, J. Mater. Chem. C, 2014, 118, 2857–2866 CAS.
- L. Stagi, D. Chiriu, A. Ardu, C. Cannas, C. M. Carbonaro and P. C. Ricci, J. Appl. Phys., 2015, 118, 125502 CrossRef.
- J. Chen, Y.-g. Liu, L. Mei, Z. Wang, M. Fang and Z. Huang, J. Mater. Chem. C, 2015, 3, 5516–5523 RSC.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00586a |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Karel
Folens
*a,
Karel
Folens