
Mesoporous graphitic carbon nitride (g-C3N4) nanosheets synthesized from carbonated beverage-reformed commercial melamine for enhanced photocatalytic hydrogen evolution†
Shaodong
Sun
 *a,
Xufeng
Gou
a,
Shasha
Tao
a,
Jie
Cui
a,
Jia
Li
a,
Qing
Yang
a,
Shuhua
Liang
*a and
Zhimao
Yang
b
*a,
Xufeng
Gou
a,
Shasha
Tao
a,
Jie
Cui
a,
Jia
Li
a,
Qing
Yang
a,
Shuhua
Liang
*a and
Zhimao
Yang
b
aShaanxi Province Key Laboratory for Electrical Materials and Infiltration Technology, School of Materials Science and Engineering, Xi’an University of Technology, Xi’an 710048, Shaanxi, People's Republic of China. E-mail: sdsun@xaut.edu.cn; liangsh@xaut.edu.cn
bSchool of Science, State Key Laboratory for Mechanical Behavior of Materials, MOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, Xi’an Jiaotong University, Xi’an 710049, Shaanxi, People's Republic of China
First published on 31st January 2019
Abstract
The application of templating and etching agents for the synthesis of mesoporous graphitic carbon nitride (mpg-C3N4) is not environmentally friendly, motivating attempts to develop a green and efficient strategy to construct mpg-C3N4 with improved photocatalytic performance. Herein, for the first time, we demonstrate a general carbonated beverage-assisted hydrothermally-reformed commercial melamine (MA) strategy for the synthesis of mpg-C3N4 nanosheets. Although the dosage of carbonated beverage (including Coca-Cola, Pepsi-Cola, Sprite and Fanta) is very small for the modification of MA precursors, the improvement in the photocatalytic activity of the mpg-C3N4 products is very remarkable. With the unique structural advantages for aligned energy bands and charge carrier migration, and numerous photocatalytic sites, the visible-light-driven photocatalytic hydrogen evolution rate (HER) of mpg-C3N4 nanosheets synthesized from a Coca-Cola-reformed MA precursor is 15.1 times higher than that of bulk g-C3N4, achieving an apparent quantum yield of 7.7% at 420 nm. Similarly, mpg-C3N4 nanosheets synthesized from Pepsi-Cola-, Sprite- and Fanta-reformed MA precursors also exhibit enhanced photocatalytic HERs.
1. Introduction
As a fascinating polymeric organic semiconductor, graphitic carbon nitride (g-C3N4) has been extensively investigated in the fields of photocatalytic hydrogen evolution, the photodegradation of organic dyes and the photoreduction of carbon dioxide.1–12 However, the photocatalytic activity of bulk g-C3N4 synthesized via traditional thermal polymerization methods is usually far from satisfactory, owing to the low surface area, short lifetimes of photogenerated charge carriers caused by the π–π conjugated electronic system, and unsuitable photo-redox potential.13–18 In order to enhance the photocatalytic performance of g-C3N4, various strategies, including morphological control, exfoliation, hybridization and doping, have been developed in the past decade.3,15,17,19–31 In particular, the development of various two-dimensional (2D) g-C3N4 nanosheets has been widely investigated in the field of photocatalysis.5,11,27,28 Nevertheless, the amount of photocatalytic active sites in g-C3N4 nanosheets is still limited because of the confined boundaries and exposed edges. Furthermore, van der Waals attractions and π–π stacking between g-C3N4 nanosheets can lead to serious aggregation and restacking, blocking the transport of hydrogen peroxide intermediates, which generally poisons g-C3N4 and decreases the photocatalytic hydrogen production activity.18,19Mesoporous graphitic carbon nitride (mpg-C3N4) nanosheets with their large specific surface area, high porosity, low density, strikingly improved electron–phonon interactions, and enhanced electron mobility along the in-plane direction, can effectively solve the above problems.1–3 The formation of holes in-plane can not only enhance mass and photogenerated charge transfer, but it can also increase the number of exposed active sites. More importantly, the energy band structure of mpg-C3N4 can be optimized through tailoring the ratio of nitrogen to carbon atoms to suitably satisfy the thermodynamic requirements for the solar-light-driven water oxidation–reduction reaction.1–3 So far, many synthetic protocols,1–3,31 such as template-assisted strategies (including hard templating and soft templating) and template-free etching-treatments of bulk g-C3N4 (etching agents include ammonia solution, ammonia gas, strong acid and strong base), have been employed to prepare 2D mpg-C3N4 nanosheets with the desired porosity as well as an intrinsic structure that meets specific needs for photocatalytic applications. However, the use of templating and etching agents is not environmentally friendly. Therefore, the development of a green and efficient strategy for the synthesis of mpg-C3N4 nanosheets is imperative.
Recently, Huang and coworkers have reported that a precursor phase-transformation strategy is effective for synthesizing three-dimensional mesoporous g-C3N4, in which the hydrothermal pretreatment of commercial melamine (MA) in aqueous urea induces a phase transformation in MA from monoclinic-phase to orthorhombic-phase.32 Similarly, the authors have also expanded this synthetic approach towards mpg-C3N4 nanosheets through calcinating thiourea-assisted hydrothermally-pretreated MA precursors.33 These mpg-C3N4 products displayed increased specific surface areas, efficient charge separation and higher photocatalytic activities compared to conventional bulk g-C3N4 obtained via the direct calcination of MA. Therefore, this precursor-reforming strategy provides a promising synthetic protocol for highly efficient g-C3N4 photocatalysts. Moreover, it has been reported that interactions between the aldehyde (–CHO) groups of glucose and the amino (–NH2) groups of MA were generated under hydrothermal conditions, which could weaken and break the chemical bonds between melon units and –NH2.34 Thus, a pre-treated MA precursor with new melon building blocks can be synthesized, which might induce the formation of novel g-C3N4 samples.
Inspired by the above considerations, for the first time, we demonstrate a general carbonated beverage-assisted hydrothermally-reformed commercial melamine (monoclinic-phase) strategy for the synthesis of mpg-C3N4 nanosheets. The involved carbonated beverages include Coca-Cola, Pepsi-Cola, Sprite and Fanta, which contain the sugar components for constructing new orthorhombic-phase MA with new melon building blocks, finally leading to the formation of mpg-C3N4 nanosheets through calcinating the reformed MA precursors in air. Although the dosage of carbonated beverages is very small for the synthesis of orthorhombic-phase MA precursors, the improvement in the photocatalytic activity of the mpg-C3N4 products is remarkable. Herein, we have firstly investigated Coca-Cola-assisted hydrothermally-reformed MA precursors for the synthesis of mpg-C3N4, which displayed much enhanced visible-light-driven photocatalytic hydrogen evolution activity, 15.1 times higher than bulk g-C3N4, and achieved an apparent quantum efficiency of 7.7% at 420 nm. Significantly, this precursor-reforming strategy could provide a general pathway to prepare mpg-C3N4 nanostructures based on an expanded carbonated beverage system. For example, mpg-C3N4 nanosheets synthesized from Pepsi-Cola-, Sprite- and Fanta-assisted hydrothermally-reformed MA precursors also exhibited higher photocatalytic hydrogen evolution performances, 13.6, 9.1 and 8.0 times than bulk g-C3N4, respectively.
2. Experimental section
2.1 Synthesis
All reagents used in experiments were of analytical grade and used without further purification. Typical mesoporous g-C3N4 nanosheets (denoted as PCN) were prepared as follows: a mixture of MA (10.0 g), Coca-Cola (0.5 mL) and deionized water (60 mL) was sonicated for 5 min at room temperature. Subsequently, the mixed solution was sealed in a 100 mL Teflon-lined autoclave and maintained at 200 °C for 12 h. Then, the autoclave was naturally cooled to room temperature. Afterwards, the as-prepared solids were washed with distilled water and absolute ethanol several times, and then dried at 60 °C for 12 h. The precursor (3.0 g) was further calcined at 550 °C for 4 h in air with a ramp rate of 0.5 °C min−1, and finally cooled naturally to room temperature to fabricate the as-synthesized PCN sample. For comparison, an HCN precursor was synthesized through a hydrothermal process in the absence of carbonated beverage under otherwise the same synthetic conditions. Then, the HCN sample was synthesized via heating the HCN precursor at 550 °C for 4 h in air. A pristine g-C3N4 sample (denoted as CN) was synthesized by directly heating MA at 550 °C for 4 h in air. In addition, the products synthesized from Pepsi-Cola-, Fanta- and Sprite-treated MA under otherwise the same synthetic conditions are denoted as CNPepsi-Cola, CNFanta and CNSprite, respectively.2.2 Characterization
Powder X-ray diffraction (XRD) patterns were obtained with a Bruker D8 Advance X-ray diffractometer, using Cu Kα1 radiation at room temperature with an accelerating voltage and current of 40 kV and 40 mA, respectively. Fourier transform infrared (FTIR) spectra were recorded using a Bruker Tensor 27 spectrometer. Elemental analysis data were collected using Eurovextor EA3000 apparatus. X-ray photoelectron spectroscopy (XPS) data were recorded using a Thermo ESCALAB Xi+ instrument with a monochromatized Al Kα line source (200 W). Transmission electron microscopy (TEM) images were obtained using a JEOL (JEM-2100) instrument at an accelerating voltage of 200 kV. Scanning electron microscopy (SEM) images were obtained by using a JEOL (JSM-7000F) instrument at an accelerating voltage of 15 kV. N2 adsorption–desorption curves were collected at 77 K using a Micromeritics ASAP 2000 Plus analyzer. Diffuse reflection spectra (DRS) were recorded using a UV/vis/NIR spectrophotometer (Hitachi U-4100) equipped with an integrating sphere in the 200–800 nm range (BaSO4 was selected as a white standard). Room temperature fluorescence emission spectra were recorded with a fluorescence spectrophotometer (FluoroLog-3, HORIBA, Jobin Yvon). Thermogravimetric curves were obtained using a NETZSCH STA449F3 instrument under a flowing argon atmosphere (flow rate: 60 mL min−1) at a heating rate of 10 °C min−1. Electron paramagnetic resonance (EPR) spectra were obtained with an EPR spectrometer (Miniscope MS 5000), using the radical scavenger dimethylpyridine N-oxide (DMPO).2.3 Electrochemical and photoelectrochemical tests
Electrochemical and photoelectrochemical measurements were carried out using a Shanghai Chenhua electrochemical system (CHI660E) under visible-light irradiation from a Xe lamp (300 W) coupled with a UV cut-off filter (λ > 420 nm), using a conventional three-electrode cell. The counter and reference electrodes were Pt wire and a saturated calomel electrode, respectively. The electrolyte solution was 0.5 M Na2SO4 (60 mL). The working electrodes were prepared as follows: 0.01 g of g-C3N4 sample was mixed with 3 mL of isopropanol, and 1 mL of distilled water was added to make slurry. The slurry was then injected onto 1.0 cm × 2.0 cm FTO glass whose sides were protected using Scotch tape, and then the samples were dried at 60 °C for 12 h and calcined at 300 °C for 1 h.2.4 Photocatalytic tests for hydrogen production
Photocatalytic hydrogen evolution was carried out using a commercial photocatalytic hydrogen generation system (CEL-SPH2N-D9, Beijing China Education Au-light Co., Ltd, China). Typically, as-obtained g-C3N4 photocatalyst (30 mg) was dispersed in aqueous solution (50 mL) containing 10 vol% triethanolamine scavenger and 3 wt% H2PtCl6·6H2O. Before hydrogen evolution testing, the above mixed solution was irradiated by a 300 W Xe lamp equipped with a 420 nm cut-off filter to deposit the Pt co-catalyst onto the photocatalyst. Subsequently, the reactor was sealed and evacuated several times to remove any air before being further irradiated under the 300 W Xe lamp (λ > 420 nm). The temperature of the reaction solution was kept at 6 °C by a flow of cooling water during the photocatalytic reaction. The amount of hydrogen evolution was recorded with a gas chromatograph (GC7920, TCD, Beijing China Education Au-light Co., Ltd, China), using high-purity N2 as the carrier gas.The apparent quantum efficiency (AQE) for hydrogen evolution was measured using a Xe lamp with a band pass filter (λ = 420 ± 10 nm, 435 ± 10 nm, 450 ± 10 nm, 500 ± 10 nm, and 550 ± 10 nm). The AQE was calculated as per the following equation:

3. Results and discussion
The mpg-C3N4 nanosheets (denoted as PCN) were prepared via the thermal polymerization of a Coca-Cola-assisted hydrothermally-reformed MA precursor without external acid/base post-treatment, as illustrated in Fig. 1. For comparison, g-C3N4 synthesized via the direct thermal condensation of MA is denoted as CN, and g-C3N4 synthesized via heating hydrothermally-reformed MA in the absence of Coca-Cola is denoted as HCN. Previously, it has been found that a hydrothermal precursor-reforming strategy could change the crystal structure of MA precursors, leading to the fabrication of a new phase, which is beneficial for the controllable synthesis of mpg-C3N4 with improved photocatalytic hydrogen evolution activity.32,33 Herein, we demonstrate for the first time that the Coca-Cola-assisted hydrothermal treatment of MA at 200 °C for 12 h can induce a phase-transformation from a monoclinic (see Fig. 2a) to an orthorhombic phase (see Fig. 2b and c), which is confirmed via X-ray diffraction (XRD) patterns collected from hydrothermally-reformed MA under different conditions (see Fig. 2). It should be highlighted that the crystal-phase of a hydrothermally-reformed MA precursor synthesized in the presence of a small amount of Coca-Cola (denoted as PCN-P) is still orthorhombic phase, except for the disappearance of some peaks owing to microstructural changes (see Fig. 2c). Furthermore, the energy dispersion spectrum (EDS) displays that oxygen, carbon and nitrogen elements are found in PCN-P (see Fig. S1, ESI†), which indicates that oxygen species might be introduced into the new hydrothermally-reformed MA precursors. Additionally, Fourier transform infrared (FTIR) spectra of the three above-mentioned precursors obviously demonstrate the formation of new molecules along with the original MA after hydrothermal treatment (see Fig. S2, ESI†). It can be obviously observed that the broad peak at 3000–3500 cm−1 resulting from amino groups is narrower and weaker for hydrothermally-treated MA, suggesting that the amino groups are partially broken during the water-assisted polymerization process.35 Similarly, the sharp peak at 750–1550 cm−1 also becomes weaker or disappears, which indicates that the triazine ring and heterocycles are broken compared with MA.35 In particular, new peaks at 1784 cm−1 (anhydride structure), 1723 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration), 1383 cm−1 (aromatic C–N stretching), 1083 cm−1 (hydroxide radicals attached to sp2 C atoms (C–OH)) and, sharply, 769 cm−1 (triazine ring) emerge compared with the MA spectrum,36–39 which confirms that oxygen-containing groups are present in the newly reformed MA molecules through a reaction between the aldehyde of sugar in the carbonated beverage and the melon unit of the original MA precursor,34 which corresponds with the EDS results (see Fig. S1, ESI†). Significantly, the morphology evolution of the three precursors from scanning electron microscopy (SEM) images can further confirm the formation of reformed microstructures. An SEM image of the original MA shows a granular shape with a compact stacking structure (see Fig. 2d and e), while the hydrothermal treated MA precursor (denoted as HCN-P) possesses an irregular shape with a smaller size compared with commercial MA (see Fig. 2f and g). However, PCN-P is a large layered structure (see Fig. 2h), and a high-magnification SEM image displays a highly ordered stacked aggregate with oriented columnar nanoparticles (see Fig. 2i). Therefore, the chemical composition and morphology of PCN-P are different from commercial MA and HCN-P; this directly determines the final features of the g-C3N4 products.
O stretching vibration), 1383 cm−1 (aromatic C–N stretching), 1083 cm−1 (hydroxide radicals attached to sp2 C atoms (C–OH)) and, sharply, 769 cm−1 (triazine ring) emerge compared with the MA spectrum,36–39 which confirms that oxygen-containing groups are present in the newly reformed MA molecules through a reaction between the aldehyde of sugar in the carbonated beverage and the melon unit of the original MA precursor,34 which corresponds with the EDS results (see Fig. S1, ESI†). Significantly, the morphology evolution of the three precursors from scanning electron microscopy (SEM) images can further confirm the formation of reformed microstructures. An SEM image of the original MA shows a granular shape with a compact stacking structure (see Fig. 2d and e), while the hydrothermal treated MA precursor (denoted as HCN-P) possesses an irregular shape with a smaller size compared with commercial MA (see Fig. 2f and g). However, PCN-P is a large layered structure (see Fig. 2h), and a high-magnification SEM image displays a highly ordered stacked aggregate with oriented columnar nanoparticles (see Fig. 2i). Therefore, the chemical composition and morphology of PCN-P are different from commercial MA and HCN-P; this directly determines the final features of the g-C3N4 products.
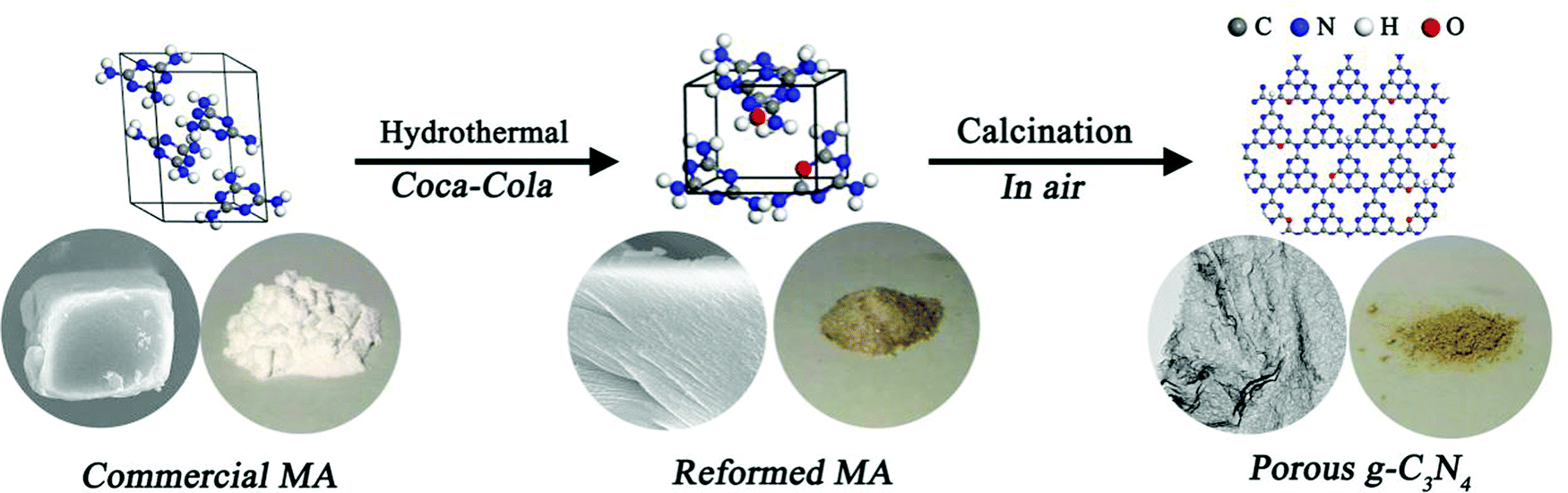 | ||
| Fig. 1 A schematic illustration of the Coca-Cola-assisted hydrothermally-modified commercial MA strategy for the synthesis of mpg-C3N4. | ||
 | ||
| Fig. 2 (a–c) XRD patterns of commercial MA, the HCN precursor and the PCN precursor, respectively. (d and e) Low-magnification and high-magnification SEM images of commercial MA. (f and g) Low-magnification and high-magnification SEM images of the HCN-precursor. (h and i) Low-magnification and high-magnification SEM images of the PCN-precursor. | ||
In this work, a transmission electron microscopy (TEM) image of the CN sample exhibits its bulk morphology (see Fig. 3a), and the HCN sample displays a nanosheet-type shape with an irregular porous structure (see Fig. 3b). As expected, the PCN sample shows laminar nanosheets with many holes (see Fig. 3c), and a large number of in-plane holes that are a few nanometers in diameter can be clearly seen (see Fig. 3d), which might provide active sites for photocatalytic reactions. The porous structure of the PCN sample inspires us to investigate detailed evolution information regarding the Brunauer–Emmett–Teller (BET) surface area and porosity. N2 adsorption/desorption isotherms of the above as-synthesized g-C3N4 products can be classified as type IV isotherms (see Fig. 3e). The isotherm of the PCN sample shows the highest N2 adsorption at high relative pressures (P/P0), corresponding to the existence of many mesopores (see the inset of Fig. 3e); the PCN sample also displays the highest BET surface area (37.4 m2 g−1), with an average pore diameter of 33.4 nm, which is 4.3 times higher than the CN sample. The pore information and BET surface area data are in agreement with the corresponding TEM morphologies (Fig. 3c and d), indicating that the Coca-Cola-assisted hydrothermally-reformed MA precursors play an important role in preparing mesoporous g-C3N4 nanosheets. Notably, the high specific surface area and large pore volume lead to a much larger volume for PCN with the same weight as CN and HCN (see Fig. S3, ESI†), and this might effectively promote the photocatalytic reaction kinetics via facilitating mass migration.
 | ||
| Fig. 3 (a and b) TEM images of CN and HCN samples, respectively. (c and d) Low-magnification and high-magnification TEM images of a PCN sample. (e and f) N2 adsorption/desorption isotherms and TGA curves for CN, HCN and PCN samples. | ||
To illuminate the formation mechanism of the mesoporous structure in the PCN sample, thermogravimetry analysis (TGA) was carried out to investigate the thermal condensation process. TGA curves of the above-mentioned precursors (namely MA, HCN-P and PCN-P) are shown in Fig. 3f. Unlike commercial MA, the TGA curves of both HCN-P and PCN-P display step changes because of the fast pyrolysis of the new CO and C–OH groups synthesized in the pre-hydrothermal treatment. The starting polymerization temperatures of MA, HCN-P and PCN-P are 405, 467 and 483 °C, respectively, which might be determined from the different chemical compositions and microstructures (see Fig. 2). Interestingly, the amounts of remaining products for MA, HCN-P and PCN-P after full condensation (T = 550 °C) are 25.85, 20.03 and 15.98%, respectively, which indicates that, compared with MA and HCN-P, PCN-P undergoes more mass loss and releases more NH3 and CO2 in the polymerization process, thus leading to the formation of more pores in the PCN sample.35 Finally, g-C3N4 nanosheets with mesoporous architecture are prepared via the thermal condensation of PCN-P.
The crystal structures of the as-obtained g-C3N4 products were characterized via XRD patterns (see Fig. 4a), which exhibit a typical (100) in-planar peak located at 13.3° and a (002) interlayer-stacking peak located at 27.3° (JCPDS 87-1526).32 The similar (100) and (002) diffraction peaks suggest that the main chemical skeletons of the three samples are well-maintained, but the two peaks are weaker for the PCN sample, indicating a smaller planar size.40 The chemical structures of the as-mentioned g-C3N4 products were obtained via FTIR spectra, as shown in Fig. 4b. The obvious sharp peak at 805 cm−1 is ascribed to the characteristic breathing mode of a heptazine ring,41 indicating the formation of the basic structure of g-C3N4. Several strong bands located between 1100 and 1700 cm−1 correspond to typical stretching modes of N–CN heterocycles,42 which is characteristic of s-triazine units.43 Broad peaks between 3000 and 3300 cm−1 originating from N–H stretching vibrations can obviously be seen,42,44 suggesting the hydrogenation of nitrogen atoms in the nanosheets.45 The peak positions in these regions undergo almost no shifting, indicating that g-C3N4 synthesized from the hydrothermally-reformed precursors can keep a similar main chemical structure as the bulk CN sample.
 | ||
| Fig. 4 (a) XRD patterns, (b) FTIR spectra, (c) high-resolution C 1s spectra and (d) high-resolution N 1s spectra for CN, HCN and PCN samples. | ||
To further analyze the effects of Coca-Cola treatment on the surface elemental composition of the product, X-ray photoelectron spectroscopy (XPS) and organic elemental analysis (OEA) measurements were carried out. Deconvoluted C 1s and N 1s XPS spectra from g-C3N4 samples are shown in Fig. 4c and d. The C 1s spectra are almost the same for the CN, HCN and PCN samples (see Fig. 4c). It can be found that the C 1s spectrum of the PCN sample could be deconvoluted into two peaks located at 284.8 and 288.4 eV, which are the result of graphitic carbon and sp2 C atoms in tri-s-triazine units (N–CN), respectively.28 The high-resolution N 1s XPS spectrum of the PCN sample is deconvoluted into four peaks with different binding energies (see Fig. 4d): the main signals are from sp2 pyridinic N involved in triazine rings (NC–N group) at 398.3 eV, pyrrolic/or pyridine N groups at 399.0 eV, sp3 hybridized tertiary nitrogen atoms of N–(C)3 at 400.5 eV, and amino functional groups carrying hydrogen atoms (C–NH or C–NH2 group) at 401.5 eV.46–50 Notably, OEA results (see Table S1, ESI†) reveal a C/N mass ratio of 0.573 for the PCN sample, which is almost unchanged compared with the value of 0.576 determined for the CN sample, indicating that the main C–N skeleton of g-C3N4 is almost unchanged when using the Coca-Cola-assisted hydrothermally-reformed MA precursor. The appearance of oxygen species could be from absorbed H2O on the surfaces of the samples. Additionally, the number of amino groups in the PCN sample (4.97%) is much lower than in the CN sample (8.70%), indicating more breaking of the interactions between melon units and amino groups in the PCN-precursor during thermal condensation, which corresponds with the above TGA results.
UV-vis diffuse reflectance spectra (DRS, Fig. 5a) were obtained to evaluate the band gap energies of the as-prepared CN, HCN and PCN, with analysis yielding values of 2.64, 2.68, and 2.65 eV, respectively (see the inset of Fig. 5b), suggesting quantum confinement effects in HCN and PCN samples.35 However, a red shift occurs in PCN compared with the HCN sample, suggesting that the bandgap can be narrowed by introducing more holes into g-C3N4, which is similar to what occurred in previous reports.32,51 Besides, light-harvesting is determined by band gaps; appropriate valence band (VB) and conductive band (CB) potentials for g-C3N4 that satisfy the redox potential of the photocatalytic reaction are also significant, and the photo-reduction and photo-oxidation abilities are determined by the potentials of the CB and VB, respectively.20,52,53 In order to identify the VB and CB potentials of the as-prepared g-C3N4, the valence band edges of the samples were measured via valence band XPS (VB-XPS), as shown in Fig. 5c. This displays that the VB potentials of CN, HCN and PCN are 1.94, 1.64 and 1.28 eV, respectively. It should be noted that the three samples satisfy the thermodynamic conditions for the photocatalytic splitting of water for hydrogen evolution. In particular, the conduction band minimum (CBM) of PCN is calculated to be up-shifted by 0.67 and 0.29 eV with respect to CN and HCN, respectively. Combining the results from Kubelka–Munk plots and VB-XPS, the band structures of CN, HCN and PCN are shown in Fig. 5d. The up-shifting of the CBM values of the HCN and PCN samples can be rationalized by considering the role of quantum confinement effects in widening bandgaps through opposingly shifting the VB and CB edges.53,54 Such an enlarged CBM suggests that the PCN sample has stronger redox capabilities than the CN and HCN samples, which could make photo-generated electrons more reductive and promote their photo-reduction abilities, finally leading to the optimization of the photocatalytic hydrogen reaction.
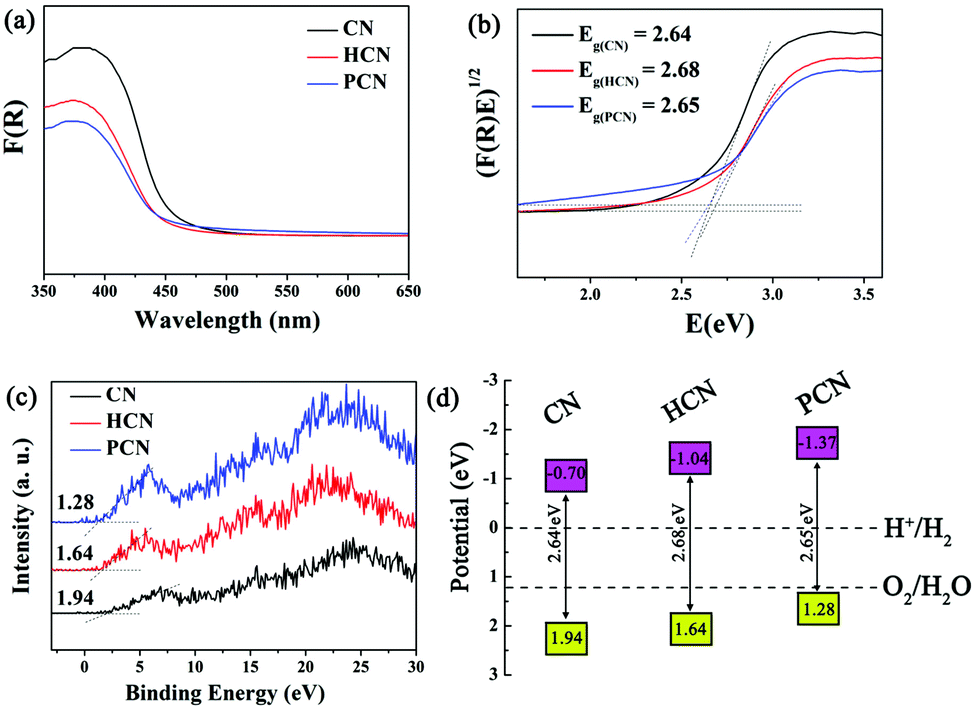 | ||
| Fig. 5 (a) UV-vis DRS, (b) plots of the transformed Kubelka–Munk function versus photon energy, (c) VB XPS spectra and (d) band structure alignments for CN, HCN and PCN samples. | ||
To elaborate on the involvement of radical species in the photocatalytic process, EPR spectra of the as-prepared CN, HCN and PCN were obtained. The ESR spectra of DMPO-trapped superoxide radicals (˙O2−) in a methanol medium and DMPO-trapped hydroxyl radicals (˙OH) in an aqueous medium have been investigated. As shown in Fig. 6a, strong signals from ˙O2− radicals are detected under visible light illumination, indicating that the photogenerated electrons in the three as-prepared samples have enough reduction ability to reduce O2 to form ˙O2− (−0.33 eV for O2/˙O2−). However, the weak oxidation potentials of the photogenerated holes in the CN, HCN and PCN samples are not sufficiently positive to oxidize water and generate ˙OH (2.69 eV for OH−/˙OH). Therefore, the observed characteristic peaks from ˙OH radicals (Fig. 6b) may be generated from ˙O2− (˙O2− + e− + 2H+ → H2O2, H2O2 + e− → ˙OH + OH−). In addition, the EPR intensities were greatly strengthened when synthesizing from the Coca-Cola hydrothermally-reformed precursor. This certifies that treatment with Coca-Cola can enhance the generation of active species, leading to much stronger electron excitability and better charge transfer properties in the PCN sample.55–57
 | ||
| Fig. 6 DMPO EPR spectra (a) in a methanol dispersion for ˙O2− and (b) in an aqueous dispersion for ˙OH were obtained under visible light irradiation for 5 min. | ||
The photocatalytic activities of the above three g-C3N4 products were evaluated via hydrogen evolution under visible light (λ > 420 nm) with 10 vol% triethanolamine as a sacrificial agent and 3 wt% Pt as a co-catalyst. As shown in Fig. 7a, the PCN sample exhibits a remarkable enhancement in photocatalytic hydrogen production compared with the CN and HCN samples. The average hydrogen evolution rate (HER) of the PCN sample is 34.8 μmol h−1 (1161.5 μmol h−1 g−1), which is 15.1 times higher than that of the CN sample (see Fig. 7b). For comparison, the porous g-C3N4 samples prepared using different carbonated beverages (such as Pepsi-Cola, Sprite and Fanta) also displayed much higher HERs compared to the CN and HCN samples under the same testing conditions (see Fig. S4, ESI†). The hydrogen evolution of a recycled PCN sample shows no serious deactivation after three cycles (see Fig. 7c). Based on TEM analyses, no substantial changes in the chemical structure of the PCN sample are observed after three successive cycles (see Fig. S5, ESI†), indicating the good stability and durability of the photocatalyst. The apparent quantum efficiency (AQE) is also calculated to understand the solar energy conversion efficiency of the PCN sample. The wavelength-dependent AQE of the PCN photocatalyst is shown in Fig. 7d; it displays an AQE of about 7.7% at 420 nm. A comparison of the photocatalytic performance for hydrogen evolution of the PCN sample with other recently reported carbon nitrides is shown in Table S2 (ESI†). It is obvious that the photocatalytic performance, especially the normalized HER, is relatively higher.
 | ||
| Fig. 7 (a) The time course of photocatalytic hydrogen evolution over 3 h for the investigated samples (30 mg) in an aqueous solution containing 10 vol% TEOA with an added 3 wt% Pt (H2PtCl6·6H2O) under visible light irradiation. (b) The corresponding HER values. (c) The recycling of a PCN sample for photocatalytic hydrogen evolution. (d) The wavelength-dependent AQE for photocatalytic hydrogen evolution over a PCN sample in the presence of Pt as a cocatalyst. | ||
As an important factor that plays a crucial part in the photocatalytic process, charge movement behavior is monitored by conducting a series of electrochemical and photoelectrochemical tests. The photogenerated electron separation activity was investigated via photocurrent tests, as shown in Fig. S6 (ESI†). They show that the photoresponses of CN, HCN and PCN are reproducible during repeated on/off cycles under visible light irradiation, suggesting efficient visible-light harvesting. Furthermore, it can be obviously seen that the PCN sample has the highest photocurrent compared to the other counterparts under visible-light irradiation, indicating much more enhanced separation efficiency of photo-induced electrons and holes. Fig. S7 (ESI†) displays electrochemical impedance spectroscopy (EIS) Nyquist plots under visible light irradiation. Compared with CN and HCN, PCN has the smallest sized arc radius. This indicates that the PCN sample has higher separation efficiency of photogenerated electron–hole pairs, corresponding to the photocurrent results.
The recombination probabilities of photogenerated carriers were investigated via photoluminescence (PL) emission spectra. Fig. S8 (ESI†) gives the PL spectra of CN, HCN and PCN samples excited at 420 nm at room temperature. Obviously, there is a significant decrease in the PL intensity of the PCN sample, compared to the CN and HCN samples. This indicates that the recombination of photogenerated electron–hole pairs can be effectively inhibited on the PCN sample.36
According to the above-mentioned results, the enhanced photocatalytic hydrogen evolution of the PCN sample can be ascribed to two reasons: namely the porous nanosheet structure and the optimization of the energy band structure by quantum confinement effects in g-C3N4. The PCN sample has a BET surface area 4.3 times that of the bulk CN sample, which provides more adsorption sites and photocatalytic reaction centers for optimizing the photocatalytic performance.16,36 Furthermore, the enlarged conductive band edge lifts the CBM of the PCN sample to a higher energy potential, which can accelerate the photo-reduction ability of electrons. Moreover, the PCN sample with its mesoporous nanosheet morphology could promote photogenerated electron migration from the bulk to the surface to participate in photocatalytic hydrogen production, leading to efficient charge generation and separation, as well as low recombination probabilities for photogenerated carriers.
4. Conclusions
In summary, a general carbonated beverage-assisted hydrothermally-reformed commercial MA (monoclinic-phase) precursor strategy has been developed for the first time to synthesize mpg-C3N4 nanosheets. Carbonated beverages (including Coca-Cola, Pepsi-Cola, Sprite and Fanta) containing sugar components played a significant role in tuning the microstructure and composition of the MA precursor from the original monoclinic phase to a new orthorhombic phase, which further influenced the thermal polymerization of the reformed MA precursors, finally leading to the formation of mesoporous nanosheets. Interestingly, although the dosage of carbonated beverage (including Coca-Cola, Pepsi-Cola, Sprite and Fanta) is very small (the volume ratio between water and the carbonated beverage is 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) during the modification of the MA precursor, the improvement in photocatalytic activity is very remarkable for the mpg-C3N4 products. For example, the visible-light-driven photocatalytic HER of a PCN sample synthesized from a Coca-Cola-reformed MA precursor is 34.8 μmol h−1 (1161.5 μmol h−1 g−1), which is 15.1 times higher than that of bulk CN, and it achieves an apparent quantum yield of 7.7% at 420 nm. The enhanced photocatalytic hydrogen evolution of the PCN sample can be attributed to its high surface area offering numerous photocatalytic sites, its enlarged conductive band edge optimizing the photo-reduction potential, and the increased number of porous holes promoting more efficient charge generation and separation, as well as the long lifetimes of photo-generated carriers. Moreover, CNPepsi-Cola, CNSprite and CNFanta samples also exhibit excellent HERs of 30.7 μmol h−1 (1023.5 μmol h−1 g−1), 20.5 μmol h−1 (683.6 μmol h−1 g−1), and 18.0 μmol h−1 (600.8 μmol h−1 g−1), respectively. This novel precursor-reforming protocol is of great significance for optimizing the chemical composition and tuning the microstructure as well as the electronic structure of g-C3N4 for improved photocatalytic hydrogen evolution.
:1) during the modification of the MA precursor, the improvement in photocatalytic activity is very remarkable for the mpg-C3N4 products. For example, the visible-light-driven photocatalytic HER of a PCN sample synthesized from a Coca-Cola-reformed MA precursor is 34.8 μmol h−1 (1161.5 μmol h−1 g−1), which is 15.1 times higher than that of bulk CN, and it achieves an apparent quantum yield of 7.7% at 420 nm. The enhanced photocatalytic hydrogen evolution of the PCN sample can be attributed to its high surface area offering numerous photocatalytic sites, its enlarged conductive band edge optimizing the photo-reduction potential, and the increased number of porous holes promoting more efficient charge generation and separation, as well as the long lifetimes of photo-generated carriers. Moreover, CNPepsi-Cola, CNSprite and CNFanta samples also exhibit excellent HERs of 30.7 μmol h−1 (1023.5 μmol h−1 g−1), 20.5 μmol h−1 (683.6 μmol h−1 g−1), and 18.0 μmol h−1 (600.8 μmol h−1 g−1), respectively. This novel precursor-reforming protocol is of great significance for optimizing the chemical composition and tuning the microstructure as well as the electronic structure of g-C3N4 for improved photocatalytic hydrogen evolution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (NSFC No. 51834009 and 51801151), the Hundred Talent Program of Shaanxi Province, the Key Laboratory Project of Shaanxi Education Department (No. 18JS070, 18JK0560 and 17JS081), the Shaanxi Province Science Fund for Distinguished Young Scholars (2018JC-027), the China Postdoctoral Science Foundation (Grant No. 2018M633643XB), the Key Research and Development Project of Shaanxi Province (No. 2017ZDXM-GY-033 and 2017ZDXM-GY-028), and the Key Laboratory Project of Science and Technology Agency (No. 13JS075).Notes and references
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- K. S. Lakhi, D. H. Park, K. Al-Bahily, W. Cha, B. Viswanathan and J. H. Choy, Chem. Soc. Rev., 2017, 46, 72–101 RSC.
- S. D. Sun and S. H. Liang, Nanoscale, 2017, 9, 10544–10578 RSC.
- G. G. Zhang, Z. A. Lan and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef PubMed.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 3183–3187 CrossRef CAS PubMed.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 124, 11984–11988 CrossRef.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- Z. Lin and X. Wang, Angew. Chem., Int. Ed., 2013, 125, 1779–1782 CrossRef.
- J. Zhang, M. Zhang, R.-Q. Sun and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed.
- J. S. Zhang, Y. Chen and X. C. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- Y. Zheng, L. H. Lin, B. Wang and X. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- Z. W. Zhao, Y. J. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- S. W. Cao, J. X. Low, J. G. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- X. P. Dong and F. X. Cheng, J. Mater. Chem. A, 2015, 3, 23642–23652 RSC.
- L. B. Jiang, X. Z. Yuan, Y. Pan, J. Liang, G. M. Zeng, Z. B. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- H. Ou, X. Chen, L. Lin, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 8729–8733 CrossRef CAS PubMed.
- M. Zhou, Z. Hou, L. Zhang, Y. Liu, Q. Gao and X. Chen, Sustainable Energy Fuels, 2017, 1, 317–323 RSC.
- J. Yan, P. Li, H. Bian, H. Wu and S. Liu, Sustainable Energy Fuels, 2017, 1, 95–102 RSC.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 9372–9376 CrossRef CAS PubMed.
- P. Yang, R. Wang, M. Zhou and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 8674–8677 CrossRef CAS PubMed.
- X.-J. Sun, D.-D. Yang, H. Dong, X.-B. Meng, J.-L. Sheng, X. Zhang, J.-Z. Wei and F.-M. Zhang, Sustainable Energy Fuels, 2018, 2, 1356–1361 RSC.
- Y. Chen, G. Jia, Y. Hu, G. Fan, Y. H. Tsang, Z. Li and Z. Zou, Sustainable Energy Fuels, 2017, 1, 1875–1898 RSC.
- P. Yang, H. Ou, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 3992–3996 CrossRef CAS PubMed.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201809897.
- S. P. Adhikari, Z. D. Hood, V. W. Chen, K. L. More, K. Senevirathne and A. Lachgar, Sustainable Energy Fuels, 2018, 2, 2507–2515 RSC.
- L. Zhang, N. Ding, J. Wu, K. Iwasaki, L. Lin, Y. Yamaguchi, Y. Shibayama, J. Shi, H. Wu, Y. Luo, K. Nakata, D. Li, X. Wang, A. Fujishima and Q. Meng, Catal. Sci. Technol., 2018, 8, 3846–3852 RSC.
- N. Tian, Y. H. Zhang, X. W. Li, K. Xiao, X. Du, F. Dong, G. I. N. Waterhouse, T. R. Zhang and H. W. Huang, Nano Energy, 2017, 38, 72–81 CrossRef CAS.
- H. W. Huang, K. Xiao, N. Tian, F. Dong, T. R. Zhang, X. Du and Y. H. Zhang, J. Mater. Chem. A, 2017, 5, 17452–17463 RSC.
- P. Zhang, X. H. Li, C. L. Shao and Y. C. Liu, J. Mater. Chem. A, 2015, 3, 3281–3284 RSC.
- C. Wang, H. Q. Fan, X. H. Ren, J. W. Ma, J. W. Fang and W. J. Wang, ChemSusChem, 2018, 11, 700–708 CrossRef CAS PubMed.
- A. I. Balabanovich, Polym. Degrad. Stab., 2004, 84, 451–458 CrossRef CAS.
- Y. J. Zhou, L. X. Zhang, W. M. Huang, Q. L. Kong, X. Q. Fan, M. Wang and J. L. Shi, Carbon, 2016, 99, 111–117 CrossRef CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- Q. Han, B. Wang, Y. Zhao, C. G. Hu and L. T. Qu, Angew. Chem., Int. Ed., 2015, 54, 1–6 CrossRef.
- H. H. Ou, L. H. Lin, Y. Zheng, P. J. Yang, Y. X. Fang and X. C. Wang, Adv. Mater., 2017, 29, 1700008 CrossRef PubMed.
- Q. Han, B. Wang, J. Gao, Z. H. Cheng, Y. Zhao, Z. P. Zhang and L. T. Qu, ACS Nano, 2016, 10, 2745–2751 CrossRef CAS PubMed.
- X. L. Wang, W. Q. Fang, H. F. Wang, H. Zhang, H. Zhao, Y. Yao and H. G. Yang, J. Mater. Chem. A, 2013, 1, 14089–14096 RSC.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 16850–16856 CrossRef CAS PubMed.
- S. B. Yang, Y. J. Gong, J. S. Zhang, L. Zhan, L. L. Ma, Z. Y. Fang, R. Vajtai, X. C. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- J. H. Liu, W. F. Li, L. M. Duan, X. Li, L. Ji, Z. B. Geng, K. K. Huang, L. H. Lu, L. S. Zhou, Z. R. Liu, W. Chen, L. W. Liu, S. H. Feng and Y. G. Zhang, Nano Lett., 2015, 15, 5137–5142 CrossRef CAS PubMed.
- Q. H. Liang, Z. Li, Z. H. Huang, F. Y. Kang and Q. H. Yang, Adv. Funct. Mater., 2015, 25, 6885–6892 CrossRef CAS.
- Y. Hou, Z. H. Wen, S. M. Cui, X. R. Guo and J. H. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed.
- H. J. Yu, R. Shi, Y. X. Zhao, T. Bian, Y. F. Zhao, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. R. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- G. H. Dong, K. Zhao and L. Z. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC.
- J. H. Li, B. Shen, Z. H. Hong, B. Z. Lin, B. F. Gao and Y. L. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- X. J. She, J. J. Wu, Z. J. Hong, H. Xu, Y. C. Yang, R. Vajtai, J. Lou, Y. Liu, D. L. Du, H. M. Li and P. M. Ajayan, Nano Energy, 2016, 27, 138–146 CrossRef CAS.
- S. Sun, J. Li, J. Cui, X. Gou, Q. Yang, Y. Jiang, S. Liang and Z. Yang, Int. J. Hydrogen Energy, 2019, 44, 778–787 CrossRef CAS.
- Q. Hao, X. Niu, C. Nie, S. Hao, W. Zou, J. Ge, D. Chen and W. Yao, Phys. Chem. Chem. Phys., 2016, 18, 31410–31418 RSC.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- Q. Hao, S. Hao, X. Niu, X. Li, D. Chen and H. Ding, Chin. J. Catal., 2017, 38, 278–286 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional Fig. S1–S8, Tables S1 and S2. See DOI: 10.1039/c8qm00577j |
| This journal is © the Partner Organisations 2019 |