
A highly selective fluorescent probe for real-time imaging of bacterial NAT2 and high-throughput screening of natural inhibitors for tuberculosis therapy†
Yinzhu
Jin‡
ab,
Zhenhao
Tian‡
c,
Xiangge
Tian‡
a,
Lei
Feng
*ab,
Jingnan
Cui
c and
Xiaochi
Ma
 *a
*a
aCollege of Pharmacy, Academy of Integrative Medicine, The National & Local Joint Engineering Research Center for Drug Development of Neurodegenerative Disease, Dalian Medical University, Dalian 116044, China. E-mail: maxc1978@163.com; leifeng@mail.dlut.edu.cn; Tel: +86-411-86110419
bInstitute of Functional Materials and Molecular Imaging, College of Emergency and Trauma, Hainan Medical University, Haikou, 571199, China
cState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China
First published on 12th November 2018
Abstract
N-Acetyltransferase 2 (NAT2) is a very important biologically functioning enzyme which plays a key role in the synthesis of cell walls in slowly growing mycobacteria. Thus, it has been widely considered to be a drug-target for anti-mycobacterial therapy. Herein, based on its catalytic characteristics, ARHB was developed for the real-time monitoring and assessment of the activity of NAT2 in different bacteria such as Mycobacterium, Staphylococcus aureus, Lactobacillus, and Pseudomonas aeruginosa. Moreover, using the highly sensitive probe ARHB, a potent natural inhibitor (KSB-4) of NAT2 was found which could effectively inhibit Mycobacterium tuberculosis H37Ra for the first time. Furthermore, the bioactivity of KSB-4 for treating tuberculosis was also evaluated in M. tuberculosis H37Ra, showing a minimum inhibitory concentration (MIC) of 25 μM. After treatment with KSB-4, M. tuberculosis H37Ra grew to be much shorter and its cell wall lysis was observed in scanning electron micrographs. All of the results fully demonstrated that ARHB could serve as a promising molecular tool for the rapid and real-time detection of NAT2 activity in a complex system, providing a novel high-throughput screening method for the discovery of new NAT2 inhibitors.
Introduction
Arylamine N-acetyltransferases (NATs) are xenobiotic conjugating enzymes that mediate the acetylation of arylamine, arylhydrazine, arylhydrazide and N-hydroxyarylamine acetyl acceptor substrates in the presence of the co-factor acetyl coenzyme A.1–4 They are commonly expressed in a wide range of organisms, both prokaryotes and eukaryotes. Until now, two NAT isoforms including NAT1 and NAT2 have been described in previous reports,5 and they have demonstrated very highly similar amino acid sequences,6,7 whilst exhibiting marked differences in substrate specificity.8,9 Several therapeutic drugs, such as the antibacterial drugs sulfamethoxazole (SMX) and p-aminosalicylate (PAS), are acetylated by NAT1 in vivo.10,11 However, NAT2 mainly activates and deactivates arylamine and hydrazine drugs including the front-line anti-tubercular drug isoniazid (INH).12,13 In a previous study, NAT2 was firstly identified and characterized at the structural level from Mycobacterium smegmatis,14 where one of its most important biological functions is participation in the synthesis of mycolic acid, which is an important component of the cell wall in the slow growth of mycobacteria.15,16 As is known, INH possesses outstanding anti-mycobacterial activity through the inhibition of NAT2 and then further inhibiting the synthesis of the mycolic acid, thus interfering with the formation of the cell wall.17Thus, NAT2 plays a vital role in the inactivation rate variation of anti-tubercular agents and has been widely considered to be a target for anti-mycobacterial therapy. Notably, recent studies have clearly indicated that acetylation of INH by NAT2 in Mycobacterium tuberculosis has contributed to the drug resistance.18,19 Hence, quantitative detection of the activity of NAT2 in different bacteria, especially in Mycobacterium, has become urgent for early-stage diagnosis which is very useful for clinical therapies and the rational use of some anti-tubercular drugs.20
Recently, fluorescent probes have been widely applied as a useful molecular tool to monitor the real-time activity of some important enzymes, and these mainly rely on advantages of having high sensitivity, being noninvasive, having real-time monitoring capability in living systems, as well as being applicable to high-throughput screening.21–27 Until now, some fluorescent probes have been developed for monitoring the enzymatic activity of NAT2 in cells and animals;28–31 however, the real activity of NAT2 in different bacterial has not been fully elucidated. Thus, it is very necessary to develop a specific sensor for the real-time detection of NAT2 bioactivity in different living bacteria, especially pathogens. Herein, according to the catalytic properties of NAT2, a fluorescent probe (ARHB) has been designed and synthesized for the real-time and specific measurement of bacterial NAT2, and exhibits high metabolic stability, selectivity, and low cytotoxicity in different bacteria (Scheme 1 and Fig. S1–S7, ESI†). Furthermore, ARHB could be successfully applied for detecting NAT2 activity in various bacteria and used for the high-throughput screening of natural NAT2 inhibitors from herbal medicines. Finally, an efficient inhibitor KSB-4 was found and verified to possess an excellent inhibitory effect toward M. tuberculosis H37Ra.
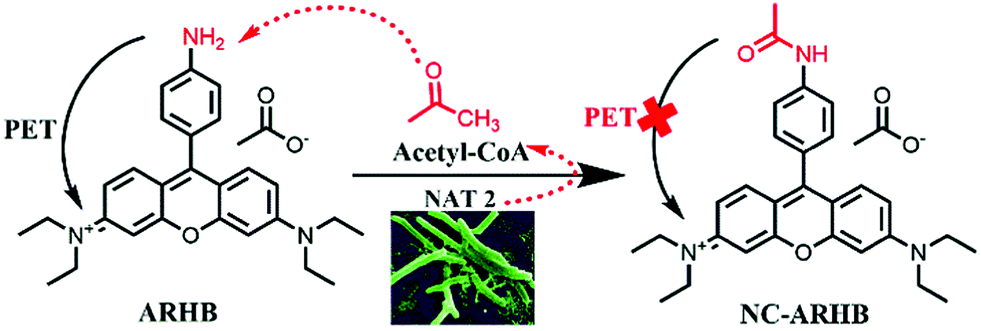 | ||
| Scheme 1 The underlying mechanism of ARHB for detecting NAT2. | ||
Experimental section
Materials and instruments
β-Galactosidase (β-Gla) and β-glucuronidase (β-GLU) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Carboxylesterases (CE) including CE1b and CE2, NAT1, NAT2, CYP1A2, CYP2C9, CYP3A4, UGT1A3, and UGT2B7 were all purchased from Corning Incorporated (NY USA); glutamate, glutamine, glycine, serine, glutathione, arginine, lysine, cysteine, tryptophan, glucose, tyrosine, bilirubin, and myristic acid were purchased from ShanghaiYuanYe (Shanghai, China). Bis-p-nitrophenylphosphate (BNPP), quercetin, and loperamide (LPA) were purchased from J&K Chemicals. Staphylococcus aureus, Pseudomonas aeruginosa, Lactobacillus, M. smegmatis MC2155 were purchased from ATCC (Manassas, VA). All fluorescence tests were performed on a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek). NMR spectra were acquired using a Bruker 501. Accurate mass detection was measured on a Fourier transform ion cyclotron resonance mass spectrometer (LTQ Orbitrap XL). The supernatants were analyzed in HPLC-UV equipment (Waters e2695 equipped with a 2998 PDA Detector). All other reagents and solvents used were of the highest grade commercially available.NMR spectra were recorded on a Bruker spectrometer using TMS as the internal standard for 1H-NMR (500 MHz) and 13C-NMR (125 MHz). High-resolution mass data were measured on a G6224A TOF mass spectrometer. Electron microscope scans were performed on an Electron Microscope QUANTA 450. The UV–vis spectra and fluorescence emission/excitation spectra were measured on a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek). A Waters 2695 system equipped with a PDA detector was used to determine ARHB and its metabolites.
Synthesis of NRHB
To a solution of 3-diethylaminophenol (1.65 g, 10.0 mmol) and 4-nitrobenzaldehyde (604.48 mg, 4.0 mmol) in 15 mL acetic acid, was added p-TsOH (34.4 mg, 0.2 mmol), and the reaction mixture was stirred at 70 °C overnight. Then the solvent was removed under reduced pressure, and the residue was dissolved in 100 mL dichloromethane (CH2Cl2), to which chloranil (738.0 mg, 3.0 mmol) was subsequently added, and the reaction mixture was stirred overnight. After removing the solvent, the crude compound was further purified by silica gel column chromatography using CH2Cl2/MeOH (20/1 v/v) as the mobile phase to afford NRHB as a dark purple solid (410.6 mg, yield: 20.4%). 1H NMR (500 MHz, CDCl3) δ 8.49 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 9.5 Hz, 2H), 6.99 (s, 2H), 6.93 (s, 2H), 3.67 (q, J = 7.0 Hz, 8H), 1.34 (t, J = 7.0 Hz, 12H). 13C NMR (125 MHz, CDCl3) δ 157.88, 155.79, 153.74, 148.95, 138.53, 131.21, 130.80, 124.22, 114.84, 112.95, 97.03, 46.36, 12.73. HRMS (ESI): [M − CH3COO−]+, calcd for 444.2282, found 444.2292 (Fig. S1–S7, ESI†).Synthesis of ARHB
A mixture of NRHB (251.6 mg, 0.5 mmol) and 10% Pd/C (26.6 mg, 0.025 mmol) in 20 mL ethanol was stirred under a hydrogen atmosphere (5 bar) for 10 h. Then the reaction mixture was filtered through Celite, and concentrated. The residue was diluted with CH2Cl2 (50 mL), and the solution was stirred overnight after the addition of chloranil (123.0 mg, 0.5 mmol). After removing the solvent, the crude compound was further purified by silica gel column chromatography using CH2Cl2/MeOH (10/1 v/v) as the mobile phase to afford ARHB as a dark red solid (56.3 mg, yield: 23.8%). 1H NMR (500 MHz, MeOD) δ 7.67 (d, J = 9.6 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 7.10 (dd, J = 9.6, 2.4 Hz, 2H), 6.94 (t, J = 5.6 Hz, 4H), 3.70 (q, J = 7.1 Hz, 8H), 1.33 (t, J = 7.1 Hz, 12H). 13C NMR (125 MHz, MeOD) δ 159.06, 158.18, 155.37, 151.29, 132.27, 131.67, 119.44, 113.87, 113.51, 112.86, 95.88, 45.26, 11.39. HRMS (ESI): [M − CH3COO−]+, calcd for 414.2540, found 414.2547.Monitoring procedure of ARHB.
To characterize the ARHB properties of NAT2, a system containing Tris–EDTA–dithiothreitol–KCl buffer (50 mM Tris–HCl, 1 mM EDTA, 10 mM DTT, 50 mM KCl), 2 μL ARHB stock solution and NAT2 (10 μg mL−1) was mixed, and after 3 min of being prewarmed at 37 °C, 200 μM acetyl-CoA generated the system. The volume of the above incubation was 0.2 mL. After 30 min of incubation at 37 °C, the reaction was quenched with 100 μL of ice-cold acetonitrile, before being vortex-mixed and centrifuged at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 20 min at 4 °C. The supernatant was subjected to analysis. Control experiments were performed in a similar way but without ARHB or NAT2, to make sure that metabolite formation was enzyme-dependent. We also explored the fluorescence spectra and linear relationship of ARHB by changing the concentrations of the enzyme or the reaction time. All assays were performed as duplicates. In order to optimize the system, we incubated the reaction at different pH values; finally, we firstly used physiological pH conditions (pH 7.4) for the assay incubation system in vitro.
000 × g for 20 min at 4 °C. The supernatant was subjected to analysis. Control experiments were performed in a similar way but without ARHB or NAT2, to make sure that metabolite formation was enzyme-dependent. We also explored the fluorescence spectra and linear relationship of ARHB by changing the concentrations of the enzyme or the reaction time. All assays were performed as duplicates. In order to optimize the system, we incubated the reaction at different pH values; finally, we firstly used physiological pH conditions (pH 7.4) for the assay incubation system in vitro.
The selectivity assays
In order to determine whether or not ARHB would be sensitive and suitable for NAT, we tested the specificity of the probe among different kinds of enzymes including oxidase CYP1A2, CYP2C9, CYP3A4, hydrolase glucuronidase, carboxylesterases 1b and 2, galactose, conjugase UGT1A1, UGT1A3, UGT2B7 and NAT1, NAT2, at a final protein concentration of 10 μg mL−1. In addition, we examined common metal ions such as Ca2+, Zn2+, Mn2+, Mg2+, Fe3+, Cu2+, K+, Ni2+, Ba2+, Na+, Sn4+ and some amino acids such as glutamate, glycine, glutamine, serine, glutathione, lysine, cysteine, tyrosine, arginine, tryptophan, glucose and endogenous substances such as myristic acid and bilirubin in our incubation system with the final concentration of 10 μM.Furthermore, different inhibitors were added to the enzymatic reaction to evaluate the fluorescence changes. The inhibitors used were as follows: BNPP (CEs general inhibitor), LPA (CE2 selective inhibitor), and quercetin, with final concentrations of 1 μM, 10 μM and 100 μM, respectively. Additionally, the IC50 values of quercetin were determined. Data were fit to a log(inhibitor) vs. normalized response-variable slope equation using GraphPad Prism 6.
Kinetic study
We conducted a kinetic study to estimate the kinetic constants of ARHB for NAT2. In brief, ARHB (0–200 μM) was incubated with NAT2 for 30 min. Both incubation times and protein concentrations were within the linear range. To calculate the kinetic parameters, we used an inhibition model: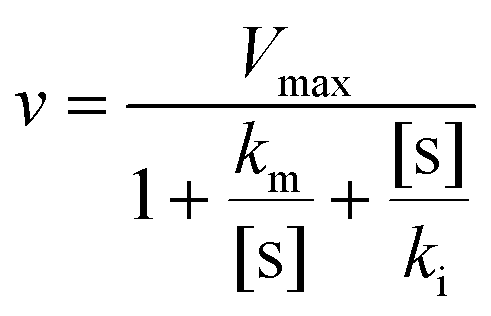 | (1) |
Fluorescence imaging in different bacterial strains
It has been reported that arylamine N-acetyltransferase activity is related to different bacteria, therefore we preformed fluorescence imaging in some bacterial strains to determine that our probe is efficient for NAT. Firstly, four bacterial strains (S. aureus, P. aeruginosa, Lactobacillus, M. smegmatis MC2155) were cultivated in Luria-Bertani (LB) liquid medium for 12 h until the optical density (OD) of the bacterial suspension was about 0.7, before the probe ARHB (20 μM) was added and incubated for 3 h at 37 °C; the bacterial cells were then harvested by centrifugation at 9000 rpm for 3 min and washed three times with PBS. The bacterial cells were then re-suspended in PBS, added dropwise on glass slides and fixed with coverslips. Bacterial imaging was carried out using laser confocal microscopy (Leica Confocal Microscope TCS SP8). Similarly, the bacterial fluids of the four bacteria strains (OD = 0.7) were diluted to 105 and cultivated in Luria-Bertani (LB) agar medium for 24 h, then the bacterial colonies were added dropwise with ARHB solution (50 μM) to react for 30 min for fluorescence measurements using the Night OWL II LB983 small animal in vivo imaging system containing a sensitive Charge Coupled Device (CCD) camera.Additionally, we cultured M. smegmatis MC2155 in both liquid and solid LB medium using the same method as mentioned above, and after pretreatment with NAT2 inhibitor quercetin (100 μM) for 15 min, the probe ARHB (50 μM) was added before incubation at 37 °C for fluorescence measurements with the Night OWL II LB983 small animal in vivo imaging system containing the sensitive CCD camera.
Screening of NAT inhibitor from natural products by using ARHB
Next, we used NAT2 as a target enzyme to screen natural products by using ARHBin vitro. In our standard incubation system, 16 natural products with a final concentration of 50 μM were added to our reaction system in the presence of ARHB (10 μM); treatment without an inhibitor was set as the control group. The same volume of DMSO as control group was added in the same incubation system. After incubation and termination as mentioned above, the supernatant was tested using a Synergy H1 microplate reader (Bio-Tek). The data were fit to a log(inhibitor) vs. normalized response-variable slope equation using GraphPad Prism.Resazurin assay
A resazurin assay was used to investigate if the compound KSB-4 had an inhibitory effect on the growth of M. tuberculosis H37Ra. The resazurin dye could indicate the cellular growth and metabolism of any bacteria by the color change from blue to pink. 50 μL portions of 7H9 broth supplemented with ADC medium (bovine serum albumin, glucose, and catalase) containing KSB-4 at a final concentration from 25 μM to 200 μM were added to 96-well plates. The M. tuberculosis H37Ra culture suspension (2 × 106 cfu mL−1), prepared by mixing the bacterial stock with the 7H9 broth supplemented ADC medium and 50 μL of culture suspension, was added into the above wells. The same volume of DMSO and culture was used as a control group in the wells. After incubating for 12 h in a 37 °C incubator, to each well was added 100 μL of 0.01% resazurin solution and the color was observed after another 3–5 h of incubation. If the resazurin color remained blue this meant that bacterial growth was inhibited. The lowest concentration of compounds that will inhibit the visible growth of a microorganism after incubation was considered as the minimum inhibitory concentration (MIC).Effects of inhibitor on morphology of M. tuberculosis H37Ra
The morphology of the M. tuberculosis H37Ra strain was observed by scanning electron microscope (SEM). The M. tuberculosis strain was inoculated in 10 mL of 7H9 liquid medium and cultured at 37 °C for 36 h, and the bacteria were collected by centrifugation at 4 °C, 3000 × g, for 5 min. The bacterial precipitate was washed using 500 μL PBS (0.1 M, pH 7.4) three times. Next, the bacteria were resuspended in 500 μL of 2.5% glutaraldehyde solution (pH 7.4). After overnight incubation, the bacteria were washed using PBS to remove the glutaraldehyde solution. Osmic acid was added to the tube ventilator to fix the bacteria for 1 h before washing with PBS as previously described. Next gradient concentrations of ethanol (50%, 70%, 80%, 90% and absolute alcohol) were added to bacteria twice for dehydration. Finally, the samples were treated with isoamyl acetate to replace the ethanol. After drying the sample using the critical point method, the samples were sputter coated with gold. All specimens were observed using a QUANTA 450 SEM.Results and discussion
Firstly, the fluorescence spectral characteristics of the ARHB response to NAT2 were elucidated, as shown in Fig. 1. After incubating with NAT2 for 30 min in the presence of the co-factor acetyl-CoA, the absorption of ARHB at 550 nm exhibited a slight increase; however, a significant enhancement of the fluorescence emission was seen at 580 nm. The fluorescence spectra were identical with NA-ARHB, additionally; a new chromatographic peak was also detected and identified as NA-ARHB (Fig. S8 and S9, ESI†), by HPLC and electrospray ionization (ESI positive) mass spectral analysis. As expected, the change of fluorescence intensity at 580 nm exhibited a good enzyme and time linearity towards NAT2 (Fig. 1 and Fig. S10, ESI†).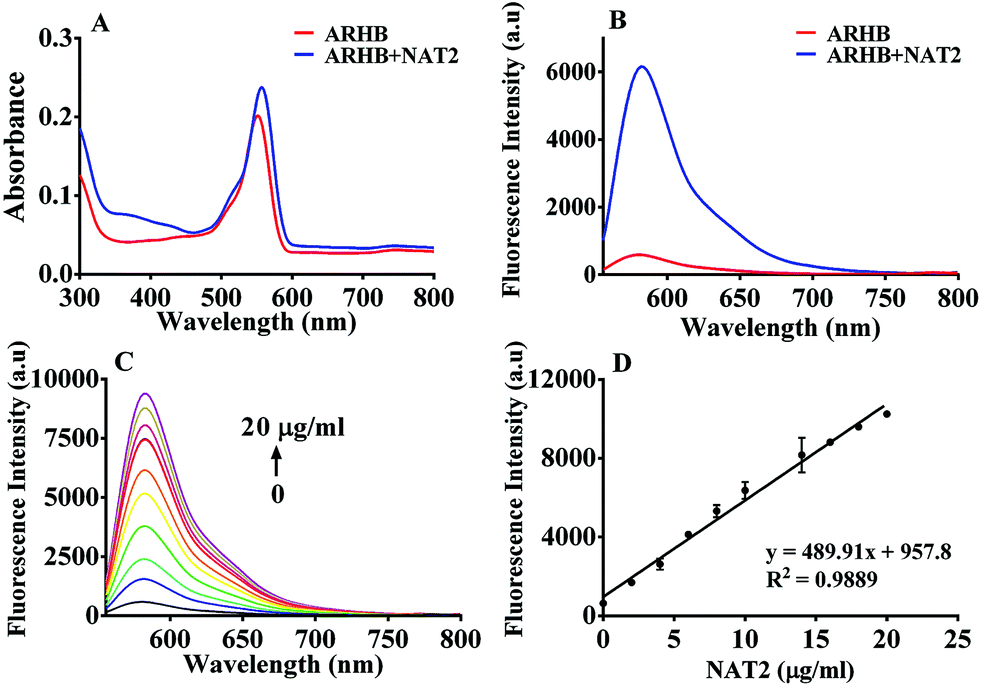 | ||
| Fig. 1 The absorption (A) and fluorescence spectra (B) of ARHB (10 μM) without (red line) and in the presence of NAT2 (blue line) in Tris–EDTA–KCl buffer (50 mM Tris–HCl, 1 mM EDTA, 50 mM KCl). (C) Fluorescence response of ARHB (10 μM) in the presence of increasing concentrations of NAT2. (D) The fluorescence intensity of ARHB at 580 nm was linearly related to the concentration of NAT2 (0–20 μg mL−1). | ||
The selectivity of ARHB towards different enzyme species was also evaluated, which is closely associated with the accurate determination of NAT2 in complex biological samples. As shown in Fig. 2A, only NAT2 could trigger the significant fluorescence enhancement at 580 nm by introducing an acetyl group to the arylamine of ARHB, whereas other enzymes including NAT1, oxidoreductases (CYP1A2, CYP3A4, CYP2C9), esterases (CE1b and CE2), hydrolases (GLU, β-Gla), and UDP-glucuronosyltransferases (UGT-1A1, -1A3, -2B7) led to negligible changes in the fluorescence intensity. In addition, different common metallic ions or endogenous substances such as amino acids and bilirubin had no influence (Fig. 2B).
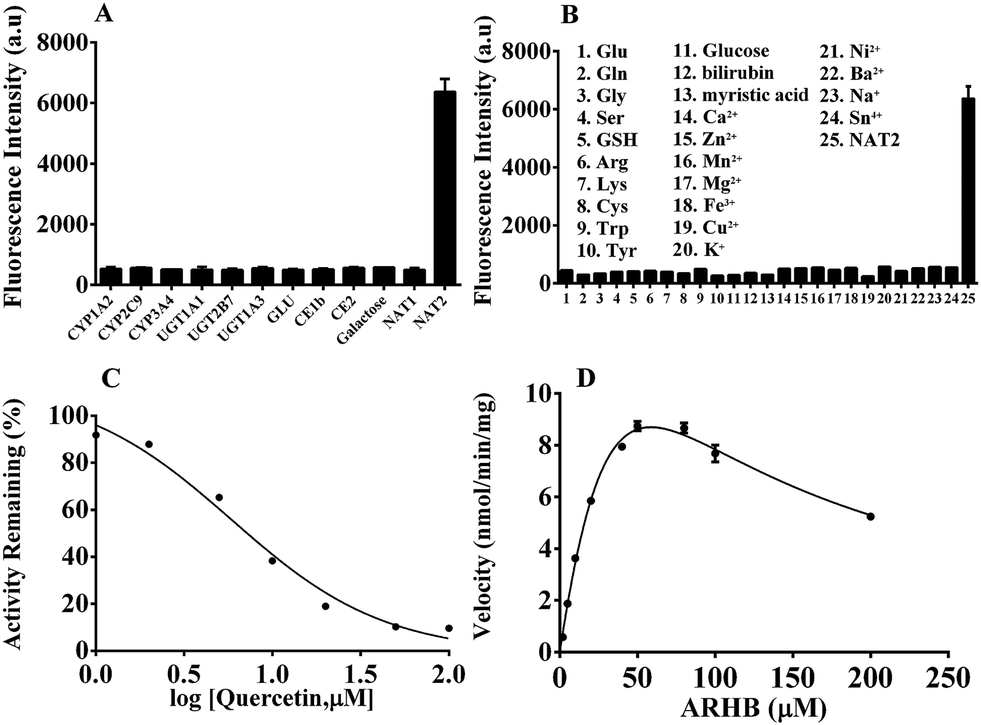 | ||
| Fig. 2 (A) Fluorescence intensity of ARHB (10 μM) in the presence of the different hydrolase sources; (B) the influence of different endogenous and exogenous substances on the fluorescence intensity; (C) chemical inhibition by quercetin with the presence of NAT2 in the incubation system; (D) the kinetic plot of ARHB hydrolysis mediated by NAT2. | ||
Furthermore, chemical inhibition was performed to confirm its selectivity, as shown in Fig. 2C. Quercetin, as a selective inhibitor for NAT2,32 could significantly block the reaction catalyzed by NAT2 with the IC50 value 7.53 μM in vitro. Other inhibitors such as BNPP (general inhibitor for CEs), and LPA (selective inhibitor for CE2) had no influence towards the fluorescence intensity mediated by NAT2. All the results fully demonstrate that ARHB could be selectively catalyzed by NAT2, and that it could serve as a highly sensitive probe for detecting the real bioactivity of NAT2 in complex biological samples. To further investigate the characteristics of the enzymatic reaction, a kinetic analysis of ARHB toward NAT2 was conducted. As shown in Fig. 2D, ARHB exhibited a substrate inhibition model towards NAT2,33 and the Km and Vmax values were calculated as 90.42 μM and 35.49 nmol min−1 mg−1, respectively. These accurate enzymatic parameters provided useful guidance in applications for measuring the activity of NAT2 in a complex biological system.
Due to the key role of NAT2 in the synthesis of mycolic acid, which is regarded as a vital component of the cell wall of bacteria, ARHB was applied for the real-time imaging of endogenous NAT2 activity in different bacteriua, including S. aureus, P. aeruginosa, Lactobacillus, and M. smegmatis. As shown in Fig. 3 and Fig. S11 (ESI†), all bacteria exhibited specific signals in red channels (570–615 nm), compared with the control group. Notably, Lactobacillus and Mycobacterium showed much stronger fluorescence intensity than S. aureus and P. aeruginosa, which clearly indicated that M. smegmatis MC2155 may possess more extensive expression of NAT2. Then, the fluorescence imaging of NAT2 in different bacterium was performed in plate medium; as shown in Fig. 3I–L, a significant fluorescence signal was detected and the intensity for different bacteria was consistent with the fluorescence imaging in bacterial thallus. All the above evidence fully demonstrated that ARHB processed good cell permeability and reaction activity with bacterial NAT2.
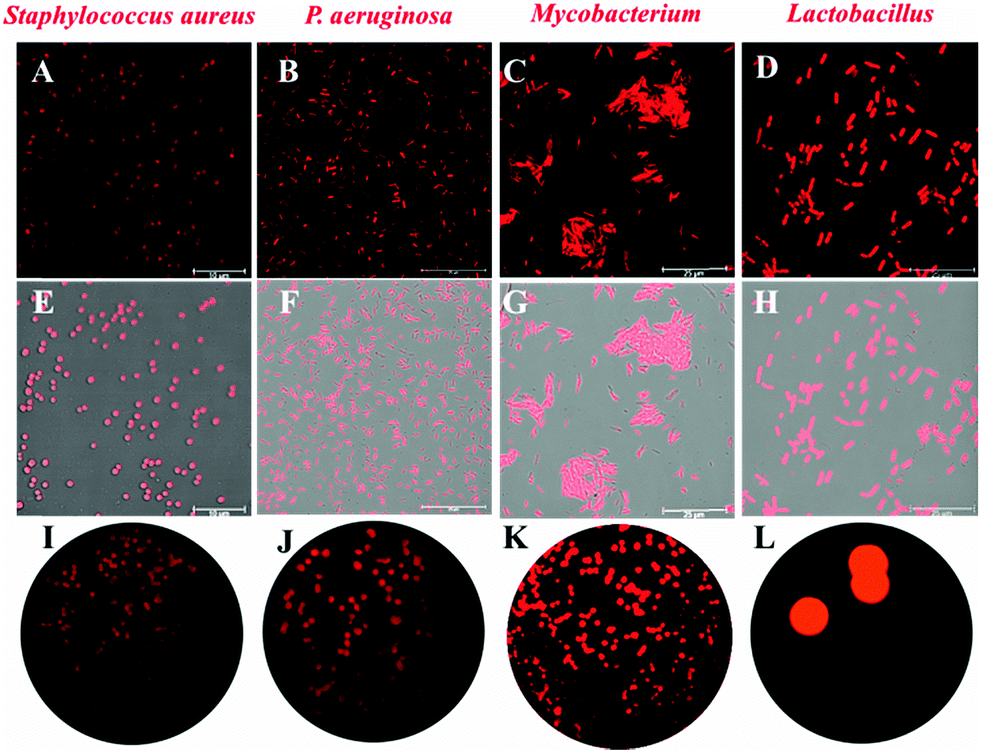 | ||
| Fig. 3 Fluorescence imaging in different bacterial strains: (A, E and I): S. aureus, (B, F and J): P. aeruginosa, (C, G and K): Lactobacillus, (D, H and L): M. smegmatis MC2155. | ||
Additionally, in order to confirm the selectivity of ARHB for NAT2 in different bacteria, an inhibition assay was performed in Mycobacterium. As shown in Fig. 4, after treatment with quercetin, a selective inhibitor of NAT2, the fluorescence intensity was significantly suppressed compared with the negative group. Importantly, the inhibition behavior was also verified by flow cytometric analysis: after incubating with ARHB (20 μM), the shift of the fluorescence signal measured in the FL2-A channel (ex: 488, em: 580 ± 25 nm), and Mycobacterium (red line) had a much larger shift than the control group (black line); furthermore, after adding the inhibitor (blue line), the shift value reduced significantly. All these results could fully indicate that ARHB was an excellent tool for imaging intracellular NAT2 in the different bacteria.
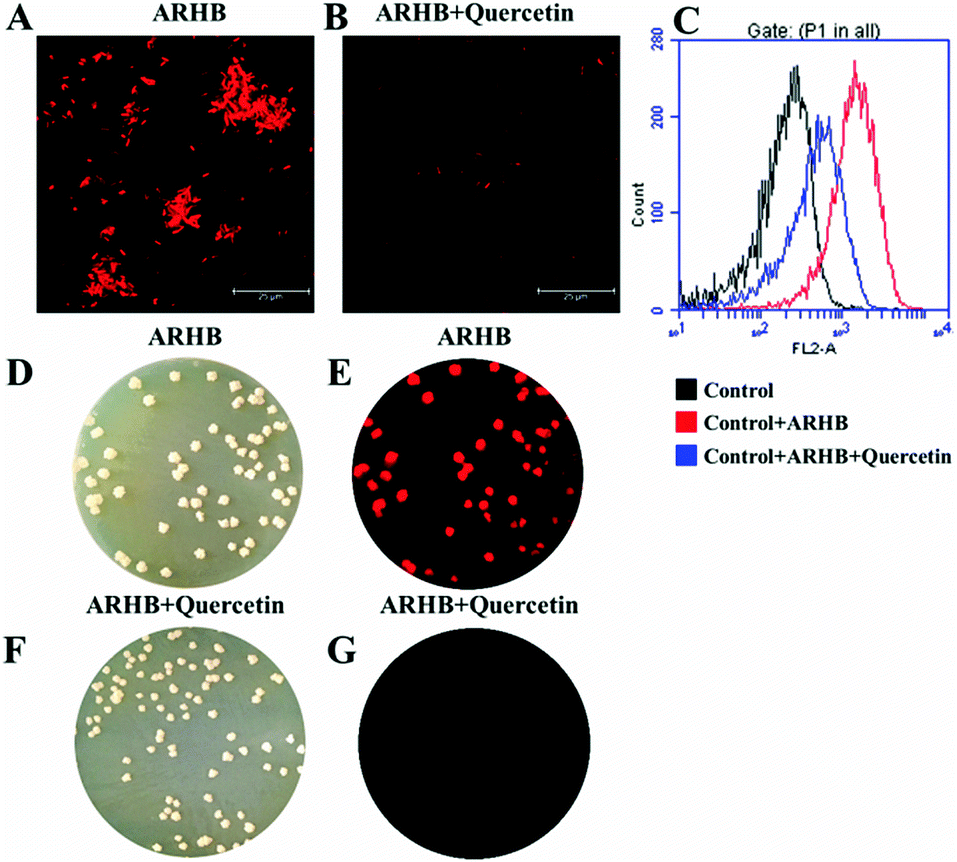 | ||
| Fig. 4 (A) Mycobacterium fluorescence imaging with 20 μM ARHB; (B) Mycobacterium was pretreated with quercetin (100 μM) for 30 min before incubating with ARHB. (C) The flow cytometric analysis of Mycobacterium incubated with ARHB with or without quercetin. Channel FL2-A: ex: 488, em: 585 ± 20 nm; (D–G) the fluorescence images of intestinal bacteria in plate medium staining with ARHB. Scale bars 25 μm. | ||
As mentioned above, NAT2 has significant role in the variation and inactivation rate of the anti-tubercular agent isoniazid, and it has been widely considered as a vital target for anti-mycobacterial therapy.34 Herein, a number of natural products and quercetin (as the positive control) were used to screen for the initial inhibition of NAT2 by ARHB (Fig. S12, ESI†). As shown in Fig. 5A, KSB-4 exhibited a significant inhibition on NAT2; however, KSD-1, KSC-3, isopsoralen, psoralen, and F-10 exhibited a middle inhibition (50% < residual activity < 80%), and KSB-2, STF-42, STF-41, STF-12, GC-21, STF-11, STF-19, naringenin, liquiritigenin, and F24 showed a slight inhibition activity (residual activity > 80%). From this, the inhibitory activity IC50 value of KSB-4 towards bacterial NAT2 was further investigated and was calculated as 10.91 μM.
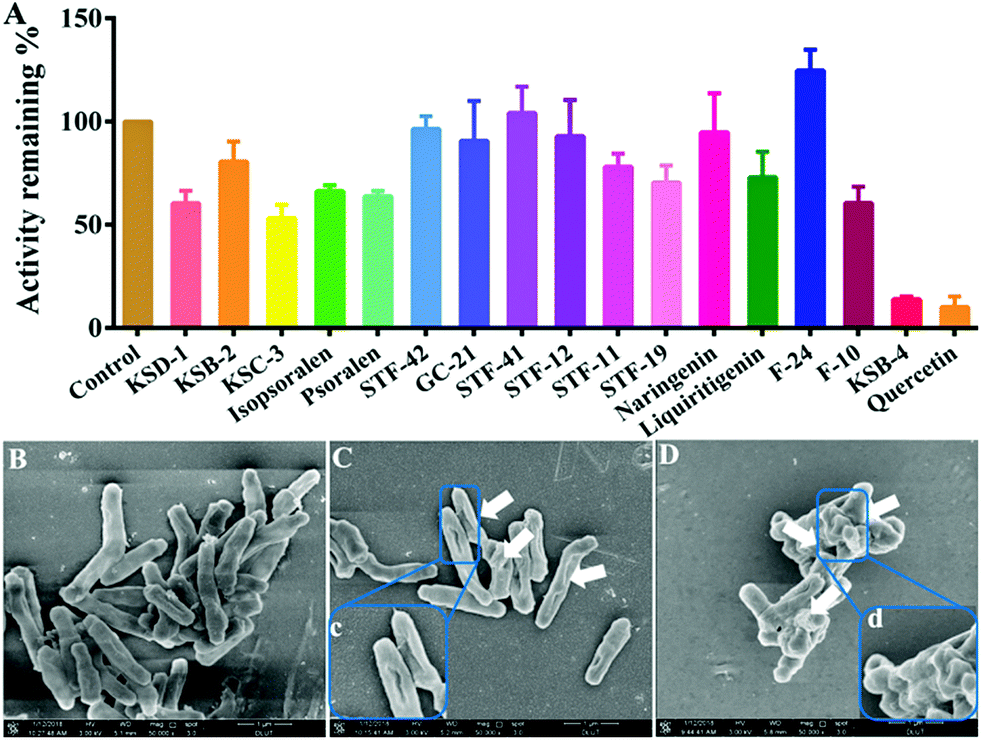 | ||
| Fig. 5 (A) Inhibition effects of 16 natural products on ARHB acetylation by NAT2; morphology of M. tuberculosis H37Ra after co-incubation with or without inhibitors. (B) Control; (C): with KSD-1; (D): with KSB-4. Scale bar: 1 μm. | ||
NAT2 plays a vital role in the synthesis of mycolic acid, which is an important component of the cell wall in the slow growing mycobacteria.35 Thus, the bioactivity of KSB-4, as a high inhibitor for bacterial NAT2, towards M. tuberculosis H37Ra was further investigated. After co-incubation of M. tuberculosis H37Ra and KSB-4 (50 μM) or KSD-1 (50 μM) for 24 h at 37 °C, the morphology of M. tuberculosis H37Ra was observed by scanning electron microscope (SEM). As shown in Fig. 5C and D, after treatment with KSB-4 and KSD-1, M. tuberculosis H37Ra became much shorter than the control group, and cell wall lysis was observed in most of the bacteria. Significant damage of the bacterial cell walls further proved the potential anti-bacterial effect of KSB-4. These results also indicated that NAT2 could be considered as a vital target for anti-mycobacterial therapy and that ARHB could serve as a useful tool for developing new potential anti-mycobacterial agents.
All of the above results demonstrated that the intracellular fluorescence change was attributed to NAT2, and that ARHB was fully capable of fluorescence imaging for endogenous NAT2 in various bacteria.
Conclusion
In summary, we have developed a fluorescent probe, termed ARHB, for detecting in various bacteria the activity of NAT2, a key enzyme in the synthesis of bacterial cell walls which has been widely considered to be a molecular target for anti-mycobacterial therapy. ARHB provides an effective high-throughput screening method for the discovery of anti-tuberculosis drugs. Also, a potent natural inhibitor of NAT2 (KSB-4) was found which could effectively inhibit M. tuberculosis H37Ra. Furthermore, the bioactivity of KSB-4 in the therapy of tuberculosis was also evaluated for M. tuberculosis H37Ra inhibition and an MIC of 25 μM was determined. After treatment with KSB-4, M. tuberculosis H37Ra grew to be much shorter and cell wall lysis was observed by SEM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 81622047, 81503201, 81473334, and 21572029), distinguished professor of Liaoning Province for financial support, and State Key Laboratory of Fine Chemicals (KF1603).References
- D. W. Hein, M. A. Doll, A. J. Fretland, M. A. Leff, S. J. Webb, G. H. Xiao, U. S. Devanaboyina, N. A. Nangju and Y. Feng, Cancer Epidemiol., Biomarkers Prev., 2000, 9, 29–42 CAS.
- L. Liu, A. Von Vett, N. Zhang, K. J. Walters, C. R. Wagner and P. E. Hanna, Chem. Res. Toxicol., 2007, 20, 1300–1308 Search PubMed.
- C. M. King, S. J. Land, R. F. Jones, M. Debiec-Rychter, M. S. Lee and C. Y. Wang, Mutat. Res., 1997, 376, 123–128 CrossRef CAS PubMed.
- S. Boukouvala and G. Fakis, Drug Metab. Rev., 2005, 37, 511–564 CrossRef CAS PubMed.
- N. Matas, P. Thygesen, M. Stacey, A. Risch and E. Sim, Cytogenet. Cell Genet., 1997, 77, 290–295 CrossRef CAS PubMed.
- N. J. Butcher, A. Arulpragasam, H. L. Goh, T. Davey and R. F. Minchin, Biochem. J., 2005, 387, 119–127 CrossRef CAS PubMed.
- W. W. Weber and D. W. Hein, Pharmacol. Rev., 1985, 37, 25–79 CAS.
- F. Pompeo, E. Brooke, A. Kawamura, A. Mushtaq and E. Sim, Pharmacogenomics, 2002, 3, 19–30 CrossRef CAS PubMed.
- D. M. Grant, M. Blum, M. Beer and U. A. Meyer, Mol. Pharmacol., 1991, 39, 184–191 CAS.
- A. E. Cribb, H. Nakamura, D. M. Grant, M. A. Miller and S. P. Spielberg, Biochem. Pharmacol., 1993, 45, 1277–1282 CrossRef CAS PubMed.
- E. Sim, A. Abuhammad and A. Ryan, Br. J. Pharmacol., 2014, 171, 2705–2725 CrossRef CAS PubMed.
- A. Toure, M. Cabral, A. Niang, C. Diop, A. Garat, L. Humbert, M. Fall, A. Diouf, F. Broly, M. Lhermitte and D. Allorge, Toxicol. Rep., 2016, 3, 826–831 CrossRef CAS PubMed.
- D. G. McCarver and R. N. Hines, J. Pharmacol. Exp. Ther., 2002, 300, 361–366 CrossRef CAS PubMed.
- E. Sim, J. Sandy, D. Evangelopoulos, E. Fullam, S. Bhakta, I. Westwood, A. Krylova, N. Lack and M. Noble, Curr. Drug Metab., 2008, 9, 510–519 CrossRef CAS PubMed.
- S. Bhakta, G. S. Besra, A. M. Upton, T. Parish, C. Sholto-Douglas-Vernon, K. J. Gibson, S. Knutton, S. Gordon, R. P. DaSilva, M. C. Anderton and E. Sim, J. Exp. Med., 2004, 199, 1191–1199 CrossRef CAS PubMed.
- I. M. Westwood, S. Bhakta, A. J. Russell, E. Fullam, M. C. Anderton, A. Kawamura, A. W. Mulvaney, R. J. Vickers, V. Bhowruth, G. S. Besra, A. Lalvani, S. G. Davies and E. Sim, Protein Cell, 2010, 1, 82–95 CrossRef CAS PubMed.
- J. Sandy, S. Holton, E. Fullam, E. Sim and M. Noble, Protein Sci., 2005, 14, 775–782 CrossRef CAS PubMed.
- M. Payton, R. Auty, R. Delgoda, M. Everett and E. Sim, J. Bacteriol., 1999, 181, 1343–1347 CAS.
- A. M. Upton, A. Mushtaq, T. C. Victor, S. L. Sampson, J. Sandy, D. M. Smith, P. V. van Helden and E. Sim, Mol. Microbiol., 2001, 42, 309–317 CrossRef CAS PubMed.
- M. Payton, C. Gifford, P. Schartau, C. Hagemeier, A. Mushtaq, S. Lucas, K. Pinter and E. Sim, Microbiology, 2001, 147, 3295–3302 CrossRef CAS PubMed.
- M. Li, Z. Liu, S. Wang, D. G. Calatayud, W. H. Zhu, T. D. James, L. Wang, B. Mao and H. N. Xiao, Chem. Commun., 2018, 54, 184–187 RSC.
- Y. Jin, X. Tian, L. Jin, Y. Cui, T. Liu, Z. Yu, X. Huo, J. Cui, C. Sun, C. Wang, J. Ning, B. Zhang, L. Feng and X. Ma, Anal. Chem., 2018, 90, 3276–3283 CrossRef CAS PubMed.
- X. Huo, X. Tian, Y. Li, L. Feng, Y. Cui, C. Wang, J. Cui, C. Sun, K. Liu and X. Ma, Sens. Actuators, B, 2018, 262, 508–515 CrossRef CAS.
- J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511–4535 RSC.
- M. Li, Z. Liu, H. C. Wang, A. C. Sedgwick, J. E. Gardiner, S. D. Bull, H. N. Xiao and T. D. James, Dyes Pigm., 2018, 149, 669–675 CrossRef CAS.
- L. Feng, Z. M. Liu, L. Xu, X. Lv, J. Ning, J. Hou, G. B. Ge, J. N. Cui and L. Yang, Chem. Commun., 2014, 50, 14519–14522 RSC.
- T. Liu, J. Ning, B. Wang, B. Dong, S. Li, X. Tian, Z. Yu, Y. Peng, C. Wang, X. Zhao, X. Huo, C. Sun, J. Cui, L. Feng and X. Ma, Anal. Chem., 2018, 90, 3965–3973 CrossRef CAS PubMed.
- X. Wang, L. Cui, N. Zhou, W. Zhu, R. Wang, X. Qian and Y. Xu, Chem. Sci., 2013, 4, 2936–2940 RSC.
- L. Cui, Y. Zhong, W. Zhu, Y. Xu and X. Qian, Chem. Commun., 2010, 46, 7121–7123 RSC.
- P. Q. Leng, F. L. Zhao, B. C. Yin and B. C. Ye, Chem. Commun., 2015, 51, 8712–8714 RSC.
- T. Terai, K. Kikuchi, Y. Urano, H. Kojima and T. Nagano, Chem. Commun., 2012, 48, 2234–2236 RSC.
- V. Kukongviriyapan, N. Phromsopha, W. Tassaneeyakul, U. Kukongviriyapan, B. Sripa, V. Hahnvajanawong and V. Bhudhisawasdi, Xenobiotica, 2006, 36, 15–28 CrossRef CAS PubMed.
- T. S. Tracy and M. A. Hummel, Drug Metab. Rev., 2004, 36, 231–242 CrossRef CAS PubMed.
- B. Chen, J. H. Li, Y. M. Xu, J. Wang and X. M. Cao, Clin. Chim. Acta, 2006, 365, 104–108 CrossRef CAS PubMed.
- S. A. Cantrell, M. D. Leavell, O. Marjanovic, A. T. Iavarone, J. A. Leary and L. W. Riley, J. Microbiol., 2013, 51, 619–626 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00514a |
| ‡ These authors equally contributed to this work. |
| This journal is © the Partner Organisations 2019 |