DOI:
10.1039/C8QM00459E
(Research Article)
Mater. Chem. Front., 2019,
3, 127-136
Enhanced efficacy of photothermal therapy by combining a semiconducting polymer with an inhibitor of a heat shock protein†
Received
12th September 2018
, Accepted 19th November 2018
First published on 20th November 2018
Abstract
Photothermal therapy (PTT) is a highly effective therapeutic modality in tumor therapy. Nevertheless, the PTT of cancer is also accompanied by thermoresistance of cells, which may alleviate the PTT efficacy or even lead to tumor recurrence. Heat shock protein 90 (HSP90) is closely related to the resistance, so a combination of inhibitors of HSP90 and photothermal agents could improve the PTT efficacy. In this work, gambogic acid (GA), an inhibitor of HSP90, is incorporated into a photothermal nanoagent, so as to reduce the influence of thermoresistance and maximize the PTT efficiency. Poly((2,5-diyl-2,3,5,6-tetrahydro-3,6-dioxo-pyrrolo(3,4-c)pyrrole-1,4-diyl)-alt-(2,2′:5′,2′′-terthiophene-5,5′′-diyl)) (PDPP3T) is selected as the robust photothermal material, and polymer-GA nanoparticles (PGNPs) incorporating both GA and PDPP3T are prepared. The PGNPs exhibit excellent photothermal activity with a high photothermal conversion efficiency of 36%, and great heating reproducibility and photoacoustic imaging performance. Besides, PGNPs are stable enough in water, in physiological conditions or under irradiation. The inhibition of HSP90 dramatically increases the apoptosis rate of cells, accordingly promoting the efficacy of PTT. Mice treated with PGNPs under 808 nm laser irradiation possess the most effective tumor inhibition, while no obvious systemic toxicity has been observed. Our study provides a valid approach to improve the PTT efficacy for more potent cancer therapy.
Introduction
Photothermal therapy (PTT), as a non-invasive therapeutic technique, has been extensively studied in recent years because of its high efficiency and specificity in tumor destruction.1–12 PTT is based on the photothermal conversion materials which convert the absorbed near-infrared (NIR, 700–1100 nm) light to heat in tumor tissues and cells, sequentially ablating the tumors.13–15 NIR light is less harmful to normal tissues and possesses deeper penetration depth than ultraviolet or visible light.16 An 808 nm laser is the most widely selected light source because of the low absorption of water in this region, which can significantly mitigate the heating effect of normal tissues.16–18 In the last decade, a myriad of organic or inorganic nanomaterials with strong absorbance in the NIR window have been extensively studied as photothermal agents (PTAs).6,11,19–35 Among them, semiconducting polymer nanoparticles (SPNs) possess high photothermal ability to convert light into heat for PTT and photoacoustic (PA) imaging.36–49 More recently, the photothermal features of SPNs have also been combined with other therapeutic methods, for instance, controlling the thermosensitive ion channels50 or enzyme activity.10
In order to ablate tumors thoroughly, harsh photothermal heating (high temperature over ∼50 °C) is required to induce complete necrosis of cells.51–54 However, due to inevitable heat diffusion, photothermal treatment at a high temperature may threaten the healthy tissues and cells nearby.55 Normally, when the temperature reaches the “hyperthermia range” (42–47 °C), tumor cells die, probably due to the degeneration of proteins.51 Nevertheless, the tumor cells are able to activate their cytoprotective pathways such as the induction of heat shock proteins (HSPs), which might rapidly repair thermal damage to proteins and lead to the thermotolerance of tumor cells upon laser irradiation.1,51,55–59 For example, HSP90, as a molecular chaperone regulating the function and stability of several signaling proteins, is closely related to antiapoptosis and tumor growth.1,60 It is reasonable that the inhibition of overexpressed HSP90 might surmount tumor thermoresistance thus improving the PTT efficacy of SPNs.1,2,7,54,61–63
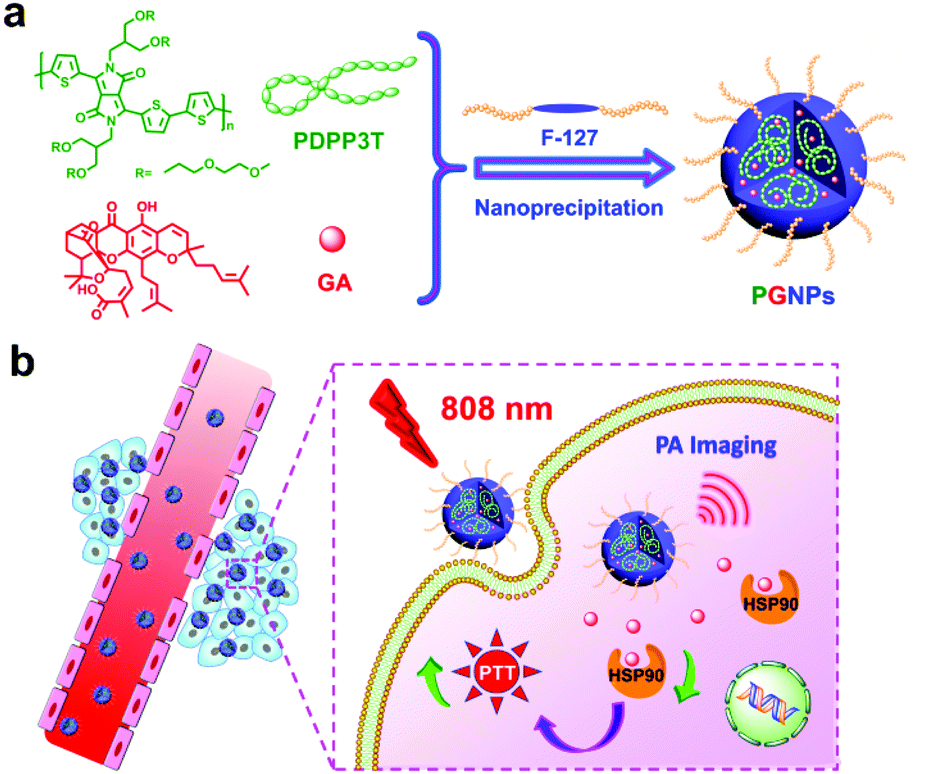
Herein, gambogic acid (GA), a natural product, was selected as the inhibitor for HSP90.64 Poly((2,5-diyl-2,3,5,6-tetrahydro-3,6-dioxo-pyrrolo(3,4-c)pyrrole-1,4-diyl)-alt-(2,2′:5′,2′′-terthiophene-5,5′′-diyl)) (PDPP3T) with branched oligo(ethylene glycol) (OEG) as the side chains and strong absorption in the NIR region was employed as the photothermal material. PDPP3T structures have already been used for photovoltaic devices,65,66 but their application in the PTT field has not been studied in detail yet. In this work, PDPP3T and GA were co-encapsulated in pluronic F-127 (F-127) to form nanoparticles (PGNPs), and the PTT potential of the PGNPs was demonstrated both in vitro and in vivo (Scheme 1). Nanoparticles without encapsulation of GA (PNPs) or PDPP3T (GNPs) were also prepared as controls. PGNPs which integrate PTA and HSP90 inhibitors could stressfully block the overexpressed HSP90 upon laser irradiation, thereby enhancing both early and late apoptosis of cells to ultimately improve the PTT efficiency.
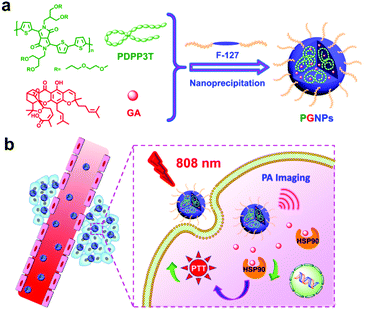 |
| | Scheme 1 Schematic illustration of (a) the preparation and (b) the enhanced PTT efficacy of PGNPs. | |
Experimental
Materials and reagents
PDPP3T was synthesized according to previous methods (yield: 50.0%).66–68 GA was purchased from Dalian Meilun Biotech Co., Ltd. F-127 was purchased from Shanghai yuanye Bio-Technology Co., Ltd. The cell viability (live-dead cell staining) assay kit and Annexin V-FITC/propidium iodide (PI) apoptosis detection kit were purchased from Jiangsu KeyGEN Biotechnology Co., Ltd. Anti-HSP90 antibody was purchased from Abcam Corporation. FITC-labeled Goat Anti-Mouse IgG (H + L) was purchased from Beyotime Biotechnology Co., Ltd. The DeadEnd™ Fluorometric TUNEL System was purchased from Promega Corporation. The other chemicals were used as obtained commercially.
Preparation of PGNPs
For the preparation of PGNPs, PDPP3T, GA and F-127 were all dissolved in 2 mL of tetrahydrofuran (THF). Then the solution was quickly dispersed into 10 mL of deionized water under sonication and sonicated for 5 min. After THF was fully evaporated, it was dialysed for 24 h. For the preparation of Nile red (NR) labeled PGNPs (NR@PGNPs), NR, PDPP3T, GA and F-127 were all dissolved in the organic phase. PNPs and GNPs were prepared with the same method in the absence of GA or PDPP3T, respectively.
Photothermal effects of PGNPs
PGNPs with different concentrations (0–5 μg mL−1) were irradiated with an 808 nm laser (1.0 W cm−2) for 10 min, and the temperature was recorded every 10 s. Besides, for a fixed concentration of PGNPs (5 μg mL−1), the influence of different power densities (0.3–1.7 W cm−2) was also recorded. The photothermal response of the PGNPs in water (5 μg mL−1) was recorded with irradiation for 10 min and the temperature during the cooling down period was also measured to determine the photothermal conversion efficiency in accordance with previous methods.69,70 The temperature variations of the PGNPs (5 μg mL−1) in water over 6 cycles of heating and natural cooling were measured to investigate their photostability.
Cellular uptake
For investigation of the cellular uptake of PGNPs by human liver hepatocellular carcinoma (HepG2) cells, NR@PGNPs were employed. Firstly, cells were seeded in 6-well culture plates with clean coverslips (1 × 105 cells per well) for 24 h. The medium (Dulbecco's modified Eagle's medium, DMEM) was then replaced with NR@PGNPs in culture medium and the cells were incubated at 37 °C for 0.5 or 2 h. After the cells were washed with phosphate-buffered saline (PBS, pH 7.4) three times, they were fixed with 4% formaldehyde. Hoechst 33258 was used to stain the nuclei. The samples were examined via confocal laser scanning microscopy (CLSM) by using a Zeiss LSM 700. Flow cytometry (FCM) was also performed to quantify the cellular uptake.
In vitro PTT efficacy
The cytotoxicity of the nanoparticles was measured via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. HepG2 cells were harvested in a logarithmic growth phase and seeded in 96-well plates (8 × 103 cells per well) followed by incubation in DMEM for 24 h. After that, cells were treated with DMEM, PNPs, GNPs or PGNPs at various concentrations for 4 h at 37 °C. Then laser irradiation (808 nm, 1.0 W cm−2, 10 min) was performed. After incubation for another 20 h, 20 μL of MTT solution (5 mg mL−1 in PBS) was added and the plates were incubated at 37 °C for another 4 h, followed by removal of the culture medium containing MTT and addition of 150 μL of dimethyl sulfoxide to each well. Finally, the absorbance of the formazan product at 490 nm was measured via a microplate reader after being shaken for 5 min.
Cell apoptosis detection assays
Cells were seeded in 6-well culture plates and pretreated with the same conditions as in MTT assays. After treatment, the cells were harvested, washed and stained with the Annexin V-FITC/PI detection kit for about 20 min. Finally, the ratio analysis of apoptosis was determined via FCM.
Live-dead cell staining
Cells were pretreated with the same conditions as in the MTT assays. After treatment, calcein-acetoxymethyl (calcein-AM)/PI were used to stain the cells for 30 min at room temperature. Finally, cell staining results were obtained by using a fluorescence microscope.
Immunofluorescence staining
Cells were seeded in 6-well culture plates with clean coverslips and pretreated with the same conditions as in the MTT assays. After laser irradiation for 1 h, the cells were firstly stained with a mouse primary anti-HSP90 antibody and then Alexa Fluor 488-labeled goat anti-mouse secondary antibody. Finally, the cells were stained with Hoechst 33258 for CLSM.
PA imaging
In vitro and in vivo PA imaging were all performed with the reported methods.71 For in vivo PA imaging, tumor-bearing nude mice were injected with PGNPs (1.5 mg kg−1) intravenously, and PA images were acquired at 0, 1, 5, 7, 12 and 24 h. The images were analyzed using ViewMOST™ software. After injection for 24 h, the mice were sacrificed, and the major organs (heart, liver, spleen, lungs and kidneys) of the mice were harvested for PA imaging.
In vivo antitumor activity and biosafety
Animal care and handling procedures were according to the guidelines of the Regional Ethics Committee for Animal Experiments. Female Kunming (KM) mice were obtained and maintained under required conditions. To evaluate antitumor effects, the animal model we used was subcutaneous hepatocarcinoma H22 tumor xenografts. The tumor-bearing mice were divided into eight groups (four in each group) randomly. They were intravenously injected with PBS, GNPs (0.15 mg kg−1 of GA), PNPs (1.5 mg kg−1 of PDPP3T) or PGNPs (1.5 mg kg−1 of PDPP3T and 0.15 mg kg−1 of GA) respectively with or without laser irradiation. After different treatments, the tumor dimensions (length and width) were measured every 2 d. Tumor volumes were calculated via length × width2/2 (mm3). 10 d later, they were all sacrificed and the tumors and main organs were collected. The main organs (heart, liver, spleen, lungs and kidneys) and tumors were fixed in 4% paraformaldehyde solution and then embedded in paraffin, sliced and stained using haematoxylin and eosin (H&E) and TUNEL.
For biosafety analysis, 4 KM mice in each group were injected with PGNPs (1.5 mg kg−1) intravenously and then irradiated with an 808 nm laser. After 10 d, the liver/kidney function makers were obtained from an automatic biochemical analyzer. Besides, the complete blood of the mice was obtained for hematology analysis.
Results and discussion
Preparation and characterization of PGNPs
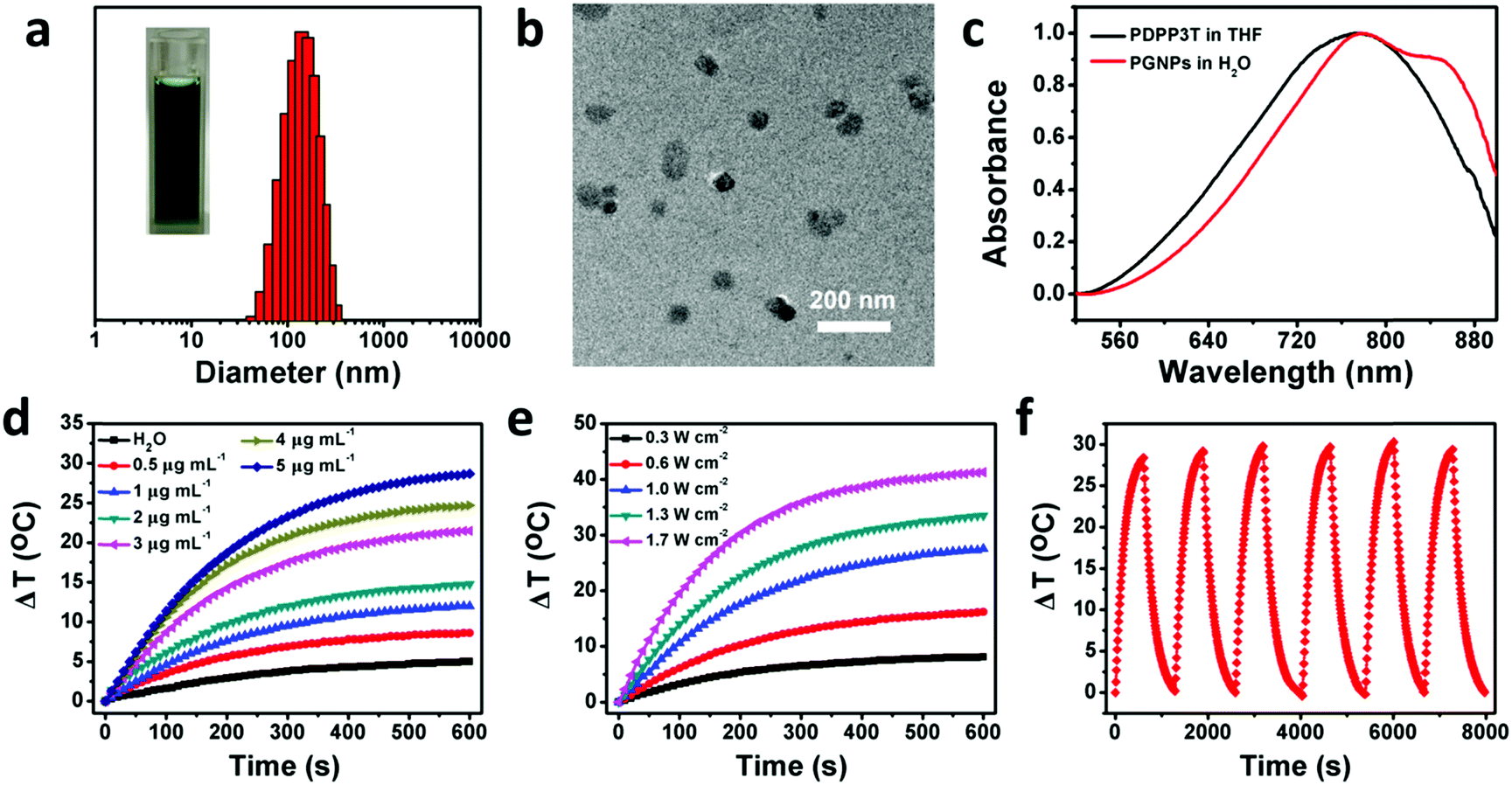
The synthetic routes of PDPP3T66 and the chemical structure of GA are shown in Scheme S1 (ESI†). The chemical structure of PDPP3T was confirmed by proton nuclear magnetic resonance (1H NMR) and elemental analysis. The number-average molecular weight (Mn) of PDPP3T is about 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. GA is not only an inhibitor of HSP90 but also a chemotherapeutic agent,72 which can inhibit the proliferation of many kinds of malignant cells, including hepatoma,73 lung cancer,74 leukemia,75 breast carcinoma76 and gastric carcinoma.77 The cytotoxicity of GA against HepG2 cells was first evaluated to determine the dosage of administration under the premise of ignoring its chemotherapeutic effect. As shown in Fig. S1 (ESI†), a maximum concentration of 0.5 μg mL−1 would be applicable. Then, PGNPs were prepared via a nanoprecipitation method. After optimizing the conditions, the loading contents were 15 wt% of PDPP3T and 1.5 wt% of GA, accompanied by loading efficiencies of 91 and 73%, respectively. The contents of PDPP3T and GA in the PGNPs were determined from the standard curves (Fig. S2 and S3, ESI†). The obtained solution of PGNPs is in dark green, and the hydrodynamic diameter of the PGNPs is 134.9 nm with a polydispersity index (PDI) of 0.15 as determined by dynamic light scattering (DLS) (Fig. 1a). The nanoparticles in the transmission electron microscopy (TEM) image (Fig. 1b) exhibit a uniform spherical morphology with sizes smaller than that measured by DLS, probably resulting from the volume shrinkage during the drying process of the TEM sample. PGNPs possess good stability both in water and PBS (pH 7.4) containing 10% fetal bovine serum (FBS) (Fig. S4, ESI†), which is of great importance for their storage and practical application. Besides, the hydrodynamic diameters of PNPs and GNPs are 148.8 and 133.8 nm with PDI of 0.172 and 0.166, respectively (Fig. S5, ESI†).
000 g mol−1. GA is not only an inhibitor of HSP90 but also a chemotherapeutic agent,72 which can inhibit the proliferation of many kinds of malignant cells, including hepatoma,73 lung cancer,74 leukemia,75 breast carcinoma76 and gastric carcinoma.77 The cytotoxicity of GA against HepG2 cells was first evaluated to determine the dosage of administration under the premise of ignoring its chemotherapeutic effect. As shown in Fig. S1 (ESI†), a maximum concentration of 0.5 μg mL−1 would be applicable. Then, PGNPs were prepared via a nanoprecipitation method. After optimizing the conditions, the loading contents were 15 wt% of PDPP3T and 1.5 wt% of GA, accompanied by loading efficiencies of 91 and 73%, respectively. The contents of PDPP3T and GA in the PGNPs were determined from the standard curves (Fig. S2 and S3, ESI†). The obtained solution of PGNPs is in dark green, and the hydrodynamic diameter of the PGNPs is 134.9 nm with a polydispersity index (PDI) of 0.15 as determined by dynamic light scattering (DLS) (Fig. 1a). The nanoparticles in the transmission electron microscopy (TEM) image (Fig. 1b) exhibit a uniform spherical morphology with sizes smaller than that measured by DLS, probably resulting from the volume shrinkage during the drying process of the TEM sample. PGNPs possess good stability both in water and PBS (pH 7.4) containing 10% fetal bovine serum (FBS) (Fig. S4, ESI†), which is of great importance for their storage and practical application. Besides, the hydrodynamic diameters of PNPs and GNPs are 148.8 and 133.8 nm with PDI of 0.172 and 0.166, respectively (Fig. S5, ESI†).
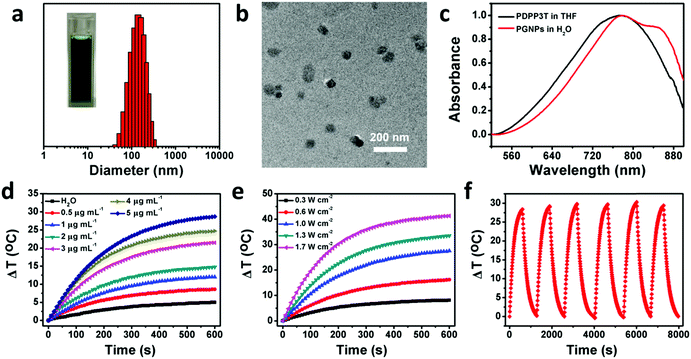 |
| | Fig. 1 Basic characterizations and photothermal performance of PGNPs. (a) DLS result (Inset: Picture of PGNPs in water) and (b) TEM image of PGNPs. (c) Absorption spectra of PDPP3T in THF and PGNPs in water. Photothermal conversion behavior of PGNPs by varying (d) concentrations (0–5 μg mL−1) and (e) laser power densities (0.3–1.7 W cm−2). (f) Heating reproducibility of PGNPs (5 μg mL−1) in water over 6 cycles of heating–cooling. | |
As shown in Fig. 1c, PGNPs in water have strong absorption in the NIR region, with a bathochromic shift relative to PDPP3T in THF, which is ascribed to the aggregation of PDPP3T molecules. The absorption coefficient of PGNPs in water at 808 nm is as high as 60.0 L g−1 cm−1, which is favorable for their photothermal application. To evaluate the photothermal performance of PGNPs in water, the temperature of the solution was recorded and compared with the variation of the concentrations and laser power. When the concentrations were increased from 0 (pure water) to 5 μg mL−1 and the power density of the 808 nm laser was fixed at 1.0 W cm−2, the increase in temperature revealed an obvious concentration-dependent tendency, as shown in Fig. 1d. The temperature elevation of the PGNPs could reach 28 °C at a low concentration of 5 μg mL−1 after laser irradiation for 10 min. A laser power-dependent temperature increase was also detected with a constant concentration of 5 μg mL−1 (Fig. 1e). The photothermal conversion efficiency of the PGNPs calculated referring to previous methods69,70 is 36% (Fig. S6, ESI†), further demonstrating their excellent photothermal performance. To validate the photothermal stability of PGNPs, 6 cycles of heating–cooling were conducted, and the elevation in temperature (Fig. 1f) and the sizes of the PGNPs (Fig. S7, ESI†) remained consistent. The high absorption coefficient, the excellent photothermal conversion behavior and the great photothermal stability of the PGNPs are beneficial to the potential application of the PGNPs in PTT.
Cellular uptake
To demonstrate the efficient cellular uptake of PGNPs by HepG2 cells, NR was selected as the fluorescent probe78 and loaded in the PGNPs (NR@PGNPs). As indicated in Fig. S8 (ESI†), the photoluminescence (PL) spectra of the PGNPs and NR@PGNPs as well as the picture of the NR@PGNPs in water substantiated the successful loading of NR. Then, CLSM was employed to observe the internalization of HepG2 cells incubated with NR@PGNPs for 0.5 and 2 h. The cell nuclei were stained with Hoechst 33258. As shown in Fig. S9a (ESI†), the fluorescence intensity of NR@PGNPs in the cells increases from 0.5 to 2 h, indicating efficient cellular uptake of PGNPs by HepG2 cells. FCM was also carried out to quantify the cellular uptake, and a time-dependent internalization of NR@PGNPs was detected (Fig. S9b, ESI†). The efficient internalization of PGNPs by cells is the firm foundation to exert their therapeutic effect.
In vitro PTT efficacy
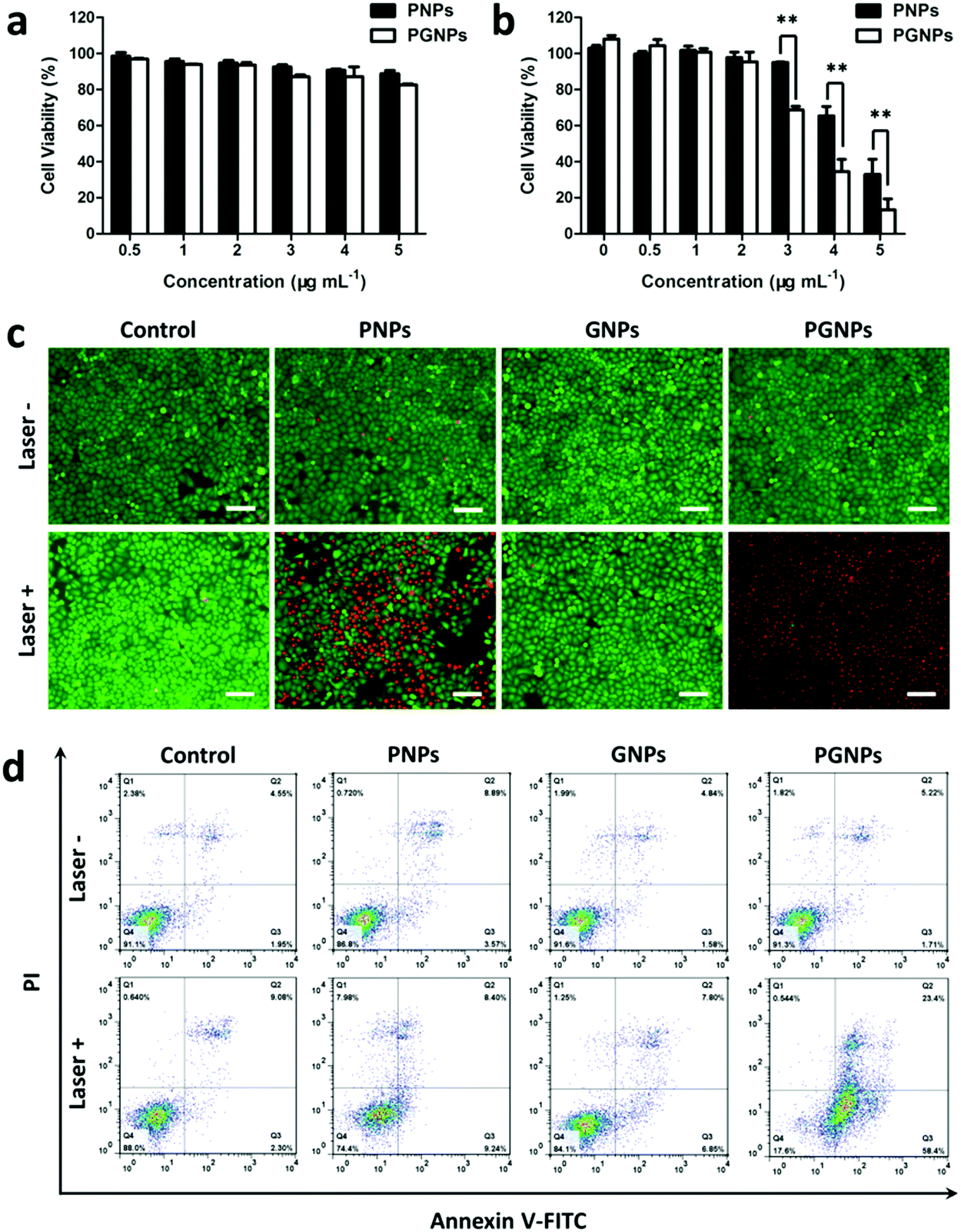
To evaluate the influence of GA on cytotoxicity, the viabilities of cells treated with PNPs or PGNPs with or without 808 nm laser irradiation were determined by MTT assays. For cells treated with PNPs or PGNPs (0–5 μg mL−1) for 24 h in the absence of laser irradiation, there was no significant cytotoxicity detected (Fig. 2a). After laser irradiation (1.0 W cm−2, 10 min), a dramatic decline in viability was observed with the increase in concentration (3–5 μg mL−1) (Fig. 2b). More importantly, the existence of GA in the PGNPs significantly decreased the viabilities of HepG2 cells relative to PNPs, with half maximal inhibitory concentration (IC50) values of 4.3 and 3.4 μg mL−1 for PNPs and PGNPs, respectively, demonstrating that the inhibition of HSP90 could enhance the PTT effect observably. However, laser irradiation makes no difference to the viability of cells treated with GNPs (Fig. S10, ESI†).
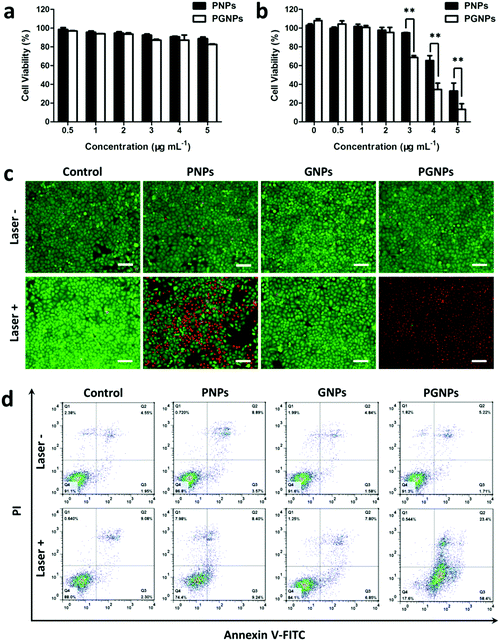 |
| | Fig. 2 Cytotoxicity against HepG2 cells. Cell viabilities of HepG2 cells treated with PNPs or PGNPs (a) without or (b) with 808 nm laser irradiation (1.0 W cm−2, 10 min). Statistical significance: **P ≤ 0.01. (c) Fluorescence images of live (green) and dead (red) HepG2 cells co-stained with Calcein-AM and PI after being incubated with DMEM, PNPs (5 μg mL−1 of PDPP3T), GNPs (0.5 μg mL−1 of GA) or PGNPs (5 μg mL−1 of PDPP3T and 0.5 μg mL−1 of GA) respectively, and then treated with or without laser irradiation (1.0 W cm−2) for 10 min. Scale bars, 100 μm. (d) FCM analysis of early and late apoptosis of HepG2 cells after the same treatments as in (c). | |
In order to visualize the in vitro PTT effect, cells were stained with calcein-AM and PI to differentiate live cells (green) and dead ones (red). Cells were treated with PBS (Control group), PNPs (5 μg mL−1 of PDPP3T), GNPs (0.5 μg mL−1 of GA) or PGNPs (5 μg mL−1 of PDPP3T and 0.5 μg mL−1 of GA), followed by laser irradiation (Laser +) or not (Laser −). Fig. 2c showed that most cells in the Control (Laser −/+), PNPs (Laser −), GNPs (Laser −/+) and PGNPs (Laser −) groups were alive with green fluorescence. Nearly all the cells in the PGNPs (Laser +) group were dead and exhibited red fluorescence, and the cell death was much more than that for the PNPs (Laser +) group (Fig. 2c), which was in line with the MTT results (Fig. 2b). Then, cell apoptosis induced by different treatments was examined quantitatively with an Annexin-V FITC/PI staining assay via FCM analysis. It could be seen from Fig. 2d that there was no obvious difference between cells treated with PBS without or with laser irradiation, indicating that the laser irradiation only has a negligible effect on cell apoptosis. The cells incubated with PNPs, GNPs or PGNPs without laser irradiation also exhibited insignificant apoptosis. For the PTT effect of PDPP3T alone, cells treated with PNPs and laser irradiation showed early and late apoptosis of 9.24% and 8.40%, respectively. In contrast, PGNPs caused the early and late apoptosis of 58.4% and 23.4%, respectively upon laser irradiation, probably due to the synergistic effect of PDPP3T and GA. The results in Fig. 2 intuitively confirmed the enhanced PTT efficacy of PGNPs with 808 nm laser irradiation by inhibiting HSP90.
The expression of HSP90
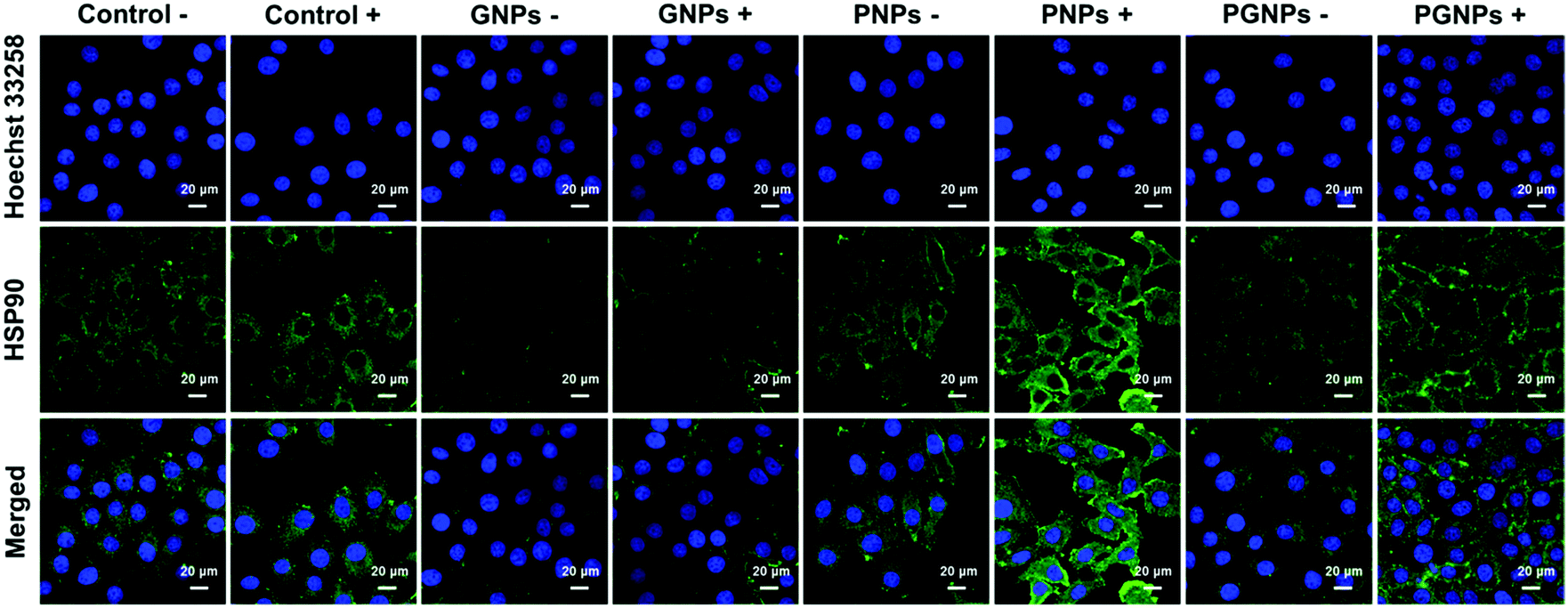
To determine the expression level of HSP90 in HepG2 cells, cells from various treatments were applied for immunofluorescence staining analysis. For conciseness, “Laser +/−” is abbreviated as “+/−”. As shown in Fig. 3 and Fig. S11 (ESI†), the expression of HSP90 was significantly upregulated in the cells incubated with PNPs after PTT treatment (PNPs +), demonstrating that PTT-induced heating stress could indeed upregulate the expression of HSP90 by cancer cells. Much lower HSP90 level was observed for cells treated with PGNPs under laser irradiation (PGNPs +) relative to that for cells in the PNPs + group, although it was higher than that for other groups, especially the GNPs −/+ groups. These results should be ascribed to the GA-mediated inhibition of the temporarily stressfully overexpressed HSP90 during PTT of cancer.
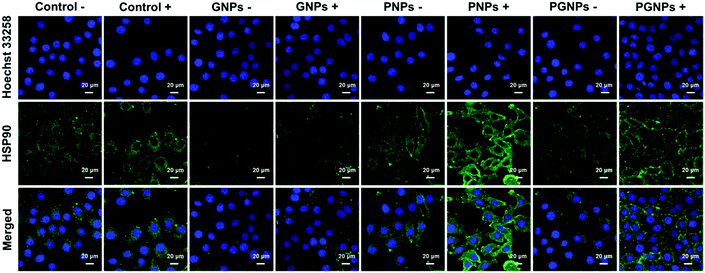 |
| | Fig. 3 The expression of HSP90 in HepG2 cells 1 h post various treatments determined by immunofluorescence staining. The HepG2 cells were treated with DMEM, GNPs, PNPs or PGNPs under 808 nm laser irradiation (1.0 W cm−2, 10 min) or not. | |
PA imaging performance of PGNPs
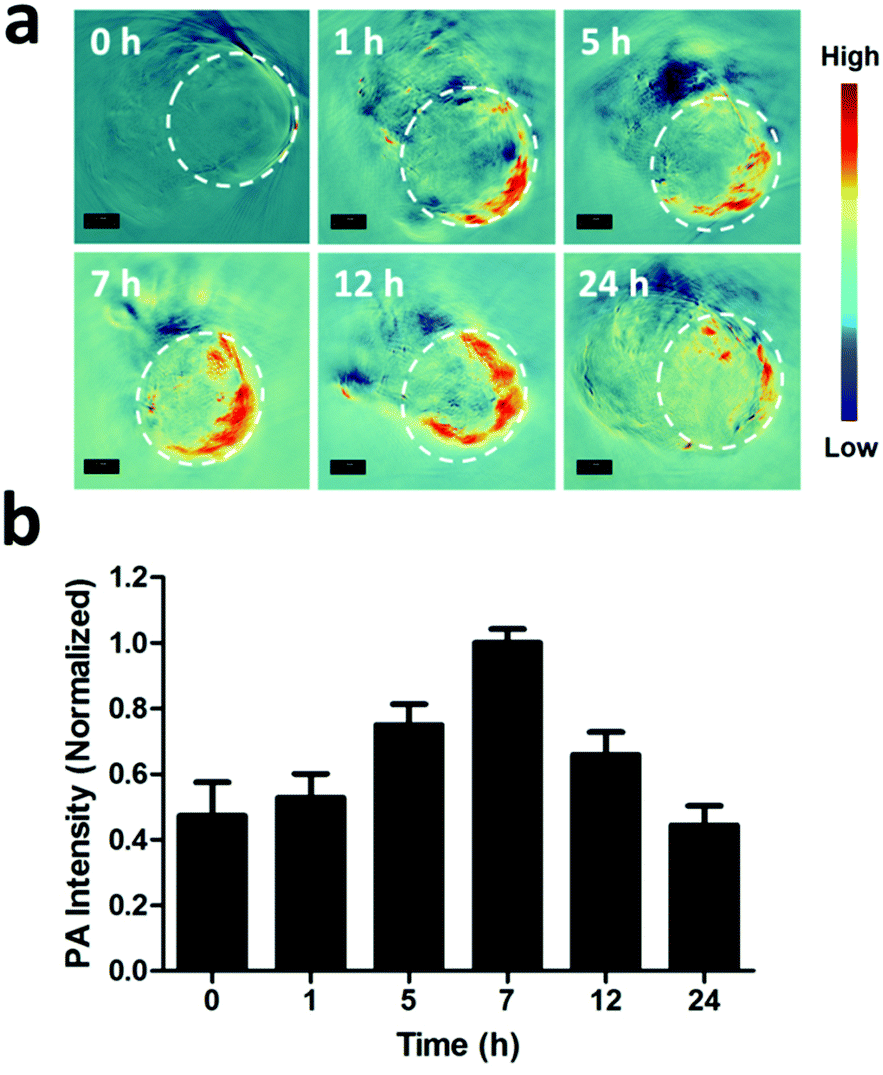
As reported, the process of photothermal transformation is accompanied by the generation of PA waves.42 Therefore, the PA imaging performance of the PGNPs was further employed to investigate the biodistribution of the nanoparticles. Firstly, the PA signals of PGNPs of varying concentrations were determined, exhibiting a linearly strengthened tendency with increasing concentration (Fig. S12, ESI†). Next, in vivo PA imaging was implemented on HepG2 tumor-bearing nude mice after intravenous injection with PGNPs (1.5 mg kg−1 of PDPP3T) for different time periods (0, 1, 5, 7, 12 and 24 h). As shown in Fig. 4, the PA signals of PGNPs in the tumor region displayed a time-dependent variation with the maximum accumulation at 7 h post injection, which was very important for determining the optimal time for laser irradiation to guarantee the best therapeutic effect. After injection for 24 h, the mice were sacrificed, and the major organs (heart, liver, spleen, lungs and kidneys) of the mice were harvested. The ex vivo PA data in Fig. S13 (ESI†) illustrate that PGNPs are mainly accumulated in the liver and spleen. These results well certified the PA imaging ability of the PGNPs, suggesting the probability of employing the nanoparticles for PA imaging-guided cancer therapy.
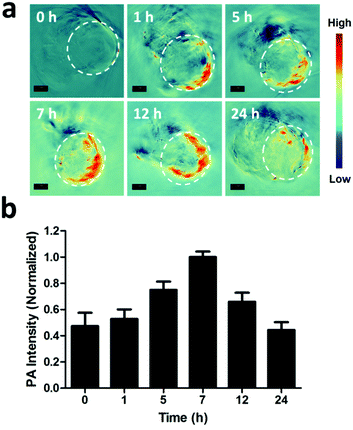 |
| | Fig. 4 (a) PA images of the tumor (in the white dashed circles) taken at specified time points after systemic administration of PGNPs (1.5 mg kg−1 of PDPP3T). Scale bar, 3 mm. (b) Quantified signal intensities of the tumor. | |
In vivo antitumor activity and biosafety
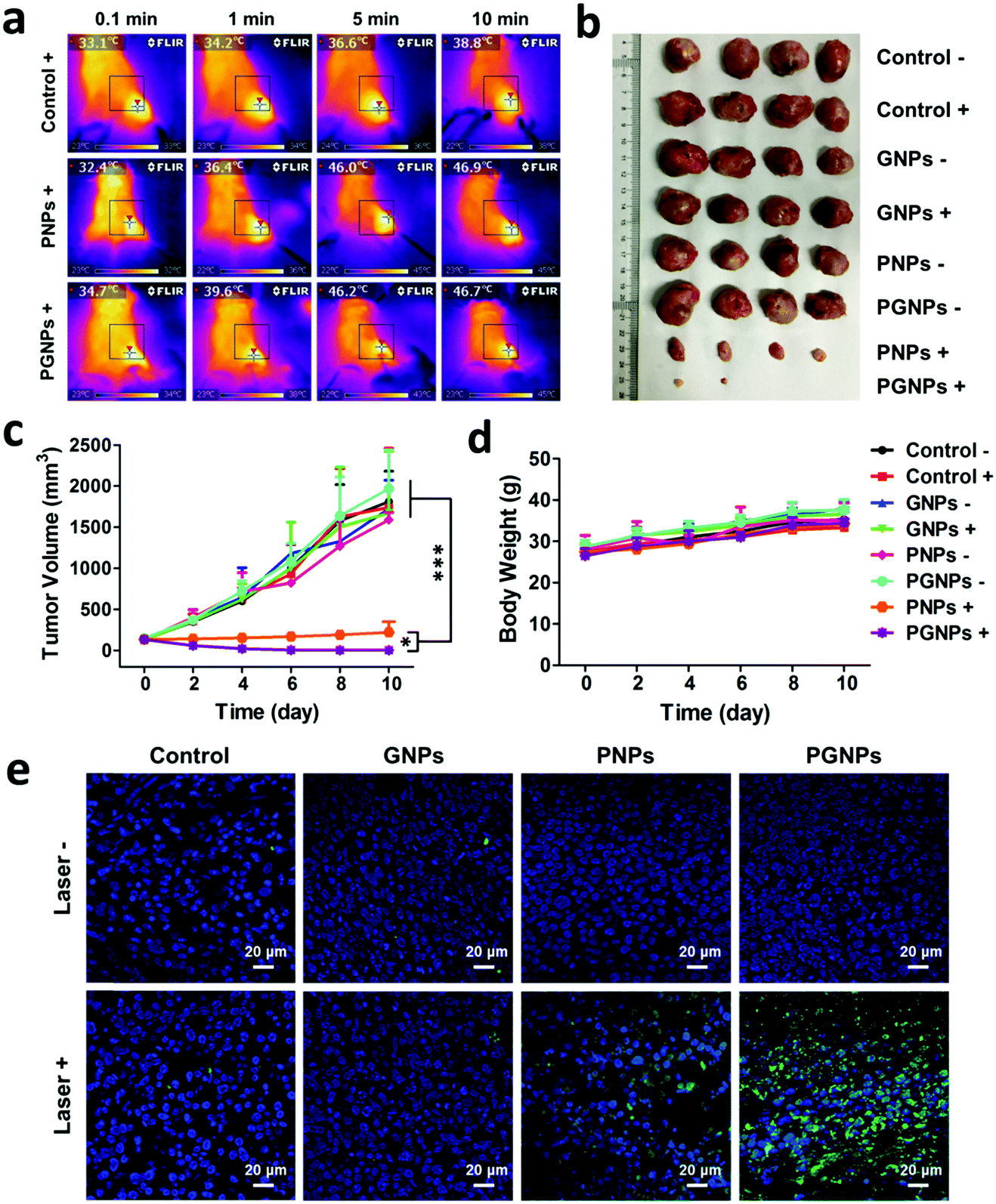
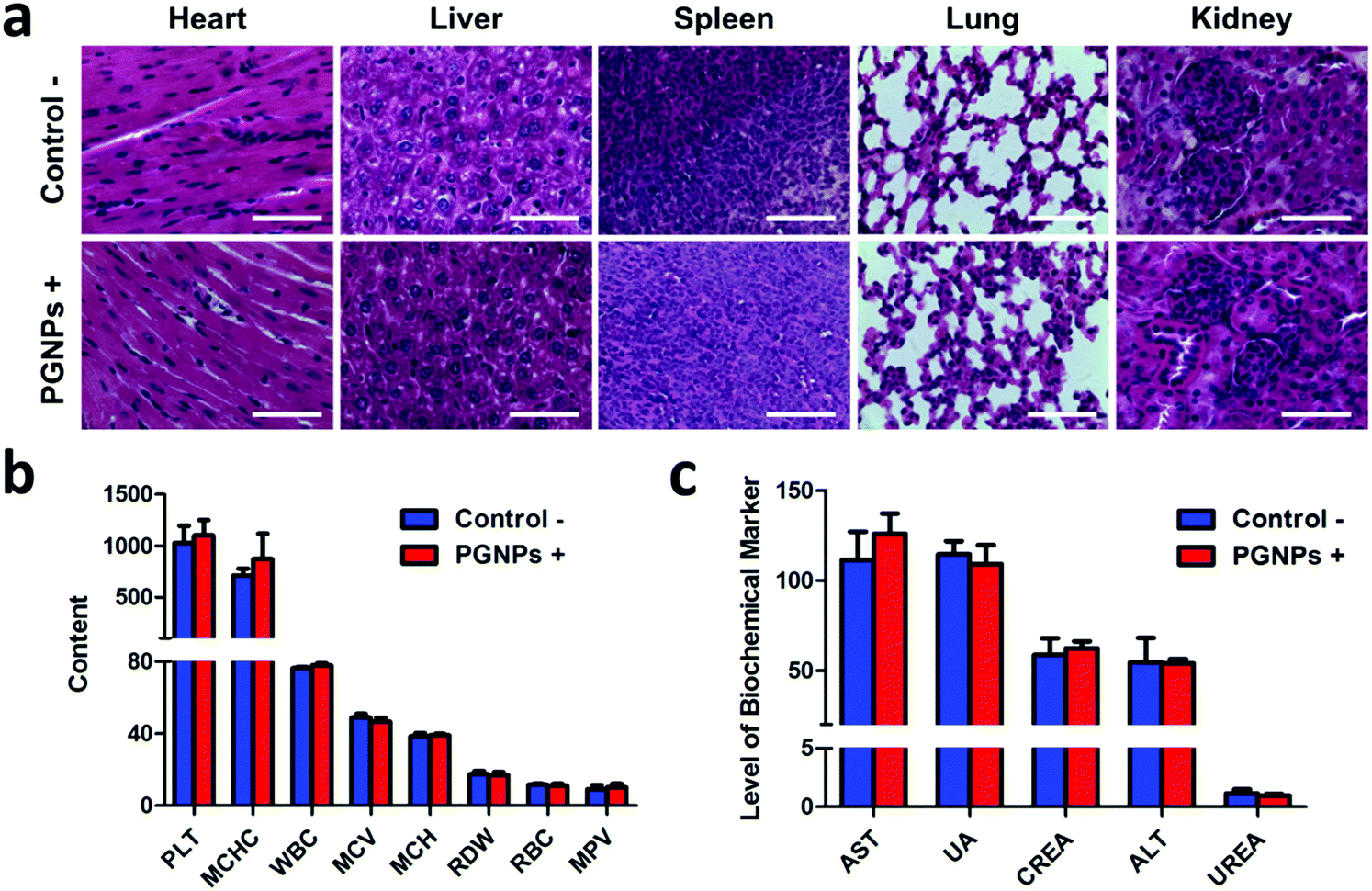
Encouraged by the enhanced cytotoxicity of PGNPs in PTT in vitro and their tumor accumulation, we then studied the in vivo PTT on H22 tumor bearing KM mice. When the tumor volumes of mice reached 100–150 mm3, they were randomly divided into eight groups the same as in the in vitro experiments: Control −/+; GNPs −/+; PNPs −/+ and PGNPs −/+. The mice were intravenously injected with PBS, GNPs (0.15 mg kg−1 of GA), PNPs (1.5 mg kg−1 of PDPP3T) or PGNPs (1.5 mg kg−1 of PDPP3T and 0.15 mg kg−1 of GA) respectively, and then they were treated with or without 808 nm laser irradiation (1.0 W cm−2, 10 min). Upon laser irradiation, an infrared (IR) thermal camera was employed to monitor the temperature changes of mice in the Control +, PNPs + and PGNPs + groups (Fig. 5a). The tumors in the Control + group showed a slight increase in temperature upon laser irradiation. The temperature of the tumors in the PNPs + and PGNPs + groups manifested a rapid rise in the first 5 min, then kept nearly unchanged between 46 and 47 °C, which validated the photothermal activity of the PNPs and PGNPs in vivo. The tumor volumes and body weight of the mice were measured every 2 d during the subsequent 10 d. The mice of the eight groups were sacrificed, and the tumors were collected at day 10. The tumor sizes in the PNPs + and PGNPs + groups were much smaller than those in other groups, and the tumors in the PGNPs + group were the smallest or even disappeared completely (Fig. 5b and Fig. S14, ESI†). As shown in Fig. 5c, the tumor volumes of the mice in the Control −/+, GNPs −/+, PNPs − and PGNPs − groups increased rapidly during the entire period. Under irradiation, PNPs could inhibit the growth of tumors upon irradiation to a large extent, but a mild increase in tumor volume was also observed. While PGNPs could decrease the volumes of all the tumors under irradiation, and some of them were even completely ablated. Next, the H&E staining of H22 tumor slices collected 2 d post treatment further verified the most severe cell damage in the PGNPs + group with that of the PNPs + group in second place, while no remarkable damage was detected in the other groups (Fig. S15, ESI†). To determine the cell apoptosis levels of tumor cells, the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining assay was carried out on tumor slices (Fig. 5e). As expected, the slice from the PGNPs + group showed the brightest green fluorescence, standing for the highest level of cell apoptosis, followed by that from the PNPs + group. However, the tumor slices from the other groups possessed a negligible level of TUNEL positive cells. It can be concluded that PGNPs are more effective than PNPs in inducing apoptosis of cancer cells under 808 nm laser irradiation in the hyperthermia range. The body weight of all the mice was in a slow upward trend (Fig. 5d), suggesting that these treatments had no significant acute toxicity to the animals. Moreover, as revealed by the H&E staining of normal tissues (heart, liver, spleen, lungs and kidneys) (Fig. 6a), the complete blood panels (Fig. 6b) and serum biochemistry (Fig. 6c), there is no obvious differences between the Control − and PGNPs + groups, indicating the great systemic safety of treatment with PGNPs and laser irradiation.
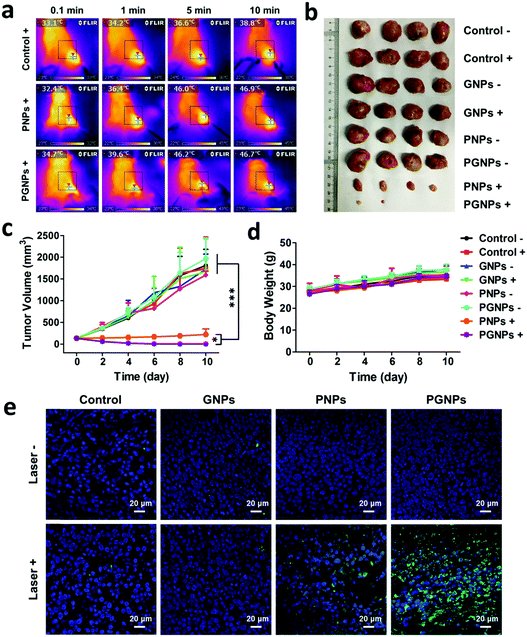 |
| | Fig. 5
In vivo therapy of tumors. (a) IR images of mice irradiated by 808 nm laser irradiation (1.0 W cm−2) after intravenous injection of PBS, PNPs or PGNPs (1.5 mg kg−1 of PDPP3T). (b) A photograph of the excised tumors from each group. (c) Growth profiles of tumors and (d) changes of body weight after different treatments. (e) TUNEL staining of tumor slices from mice subjected to various treatments 2 d later. Statistical significance: *P ≤ 0.05; ***P ≤ 0.001. | |
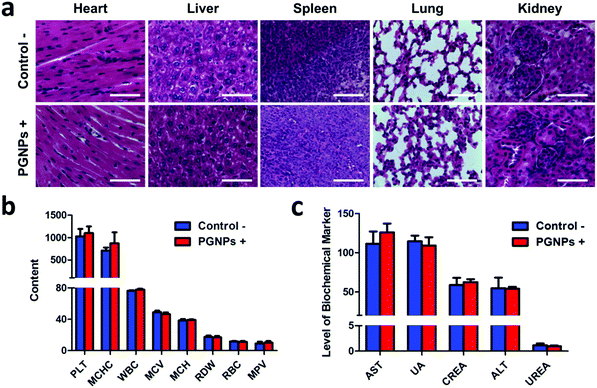 |
| | Fig. 6 (a) H&E staining of major organs (heart, liver, spleen, lungs and kidneys) of mice in the Control − and PGNPs + groups. Scale bars: 200 μm. (b) Hematology data and (c) serum biochemical analysis of kidney and liver function parameters of the mice (n = 4) in the Control – and PGNPs + groups after treatment for 10 d. PLT, platelet; MCHC, mean corpuscular hemoglobin concentration; WBC, white blood cell count; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; RDW, red cell distribution width; RBC, red blood cell; MPV, mean platelet volume; AST, aspartate aminotransferase; UA, uric acid; CREA, creatinine; ALT, alanine transaminase; and UREA, urea nitrogen. | |
Conclusions
In summary, we have constructed PGNPs integrating the photothermal properties of PDPP3T with inhibition towards HSP90 for highly potent cancer therapy. PGNPs possess a high photothermal conversion efficiency of 36% under 808 nm laser irradiation, and have excellent physiological stability as well as photostability. Furthermore, the PA imaging ability of the PGNPs makes imaging-guided cancer therapy possible. More importantly, the PGNPs could induce more severe cell apoptosis in vitro and effective inhibition of tumor growth in vivo than PNPs without the inhibitor of HSP90. This work demonstrates a new design to improve the PTT efficacy of PTAs based on semiconducting polymer nanoparticles, thus realizing more effective cancer therapy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (Project No. 51522307).
Notes and references
- H. Luo, Q. Wang, Y. Deng, T. Yang, H. Ke, H. Yang, H. He, Z. Guo, D. Yu, H. Wu and H. Chen, Adv. Funct. Mater., 2017, 27, 1702834 CrossRef.
- B.-K. Wang, X.-F. Yu, J.-H. Wang, Z.-B. Li, P.-H. Li, H. Wang, L. Song, P. K. Chu and C. Li, Biomaterials, 2016, 78, 27–39 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- X. Cai, J. Liu, W. H. Liew, Y. Duan, J. Geng, N. Thakor, K. Yao, L.-D. Liao and B. Liu, Mater. Chem. Front., 2017, 1, 1556–1562 RSC.
- M. P. Melancon, M. Zhou and C. Li, Acc. Chem. Res., 2011, 44, 947–956 CrossRef CAS PubMed.
- X. Zhen, C. Xie and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3938–3942 CrossRef CAS PubMed.
- Y. Yang, W. Zhu, Z. Dong, Y. Chao, L. Xu, M. Chen and Z. Liu, Adv. Mater., 2017, 29, 1703588 CrossRef PubMed.
- C. Wang, L. Xu, C. Liang, J. Xiang, R. Peng and Z. Liu, Adv. Mater., 2014, 26, 8154–8162 CrossRef CAS PubMed.
- P. Huang, J. Lin, W. Li, P. Rong, Z. Wang, S. Wang, X. Wang, X. Sun, M. Aronova, G. Niu, D. Leapman Richard, Z. Nie and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 13958–13964 CrossRef CAS PubMed.
- J. Li, C. Xie, J. Huang, Y. Jiang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3995–3998 CrossRef CAS PubMed.
- Q. Dong, X. Wang, X. Hu, L. Xiao, L. Zhang, L. Song, M. Xu, Y. Zou, L. Chen, Z. Chen and W. Tan, Angew. Chem., Int. Ed., 2017, 57, 177–181 CrossRef PubMed.
- Z. Yang, W. He, H. Zheng, J. Wei, P. Liu, W. Zhu, L. Lin, L. Zhang, C. Yi, Z. Xu and J. Ren, Biomaterials, 2018, 161, 1–10 CrossRef CAS PubMed.
- Q. Ban, T. Bai, X. Duan and J. Kong, Biomater. Sci., 2017, 5, 190–210 RSC.
- Y. Liu, X. Ji, J. Liu, W. L. Tong Winnie, D. Askhatova and J. Shi, Adv. Funct. Mater., 2017, 27, 1703261 CrossRef PubMed.
- W. Huang, Y. Huang, Y. You, T. Nie and T. Chen, Adv. Funct. Mater., 2017, 27, 1701388 CrossRef.
- X. Deng, K. Li, X. Cai, B. Liu, Y. Wei, K. Deng, Z. Xie, Z. Wu, P. A. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Mater., 2017, 29, 1701266 CrossRef PubMed.
- V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prud’homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS PubMed.
- W. Cheng, J. Nie, N. Gao, G. Liu, W. Tao, X. Xiao, L. Jiang, Z. Liu, X. Zeng and L. Mei, Adv. Funct. Mater., 2017, 27, 1704135 CrossRef.
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- S. Thota, Y. Wang and J. Zhao, Mater. Chem. Front., 2018, 2, 1074–1089 RSC.
- W. M. Girma, S.-H. Tzing, P.-J. Tseng, C.-C. Huang, Y.-C. Ling and J.-Y. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 4590–4602 CrossRef CAS PubMed.
- S. Zhang, Q. Huang, L. Zhang, H. Zhang, Y. Han, Q. Sun, Z. Cheng, H. Qin, S. Dou and Z. Li, Nanoscale, 2018, 10, 3130–3143 RSC.
- N. Yan, X. Wang, L. Lin, T. Song, P. Sun, H. Tian, H. Liang and X. Chen, Adv. Funct. Mater., 2018, 28, 1800490 CrossRef.
- Y. Zhan, Y. Liu, H. Zu, Y. Guo, S. Wu, H. Yang, Z. Liu, B. Lei, J. Zhuang, X. Zhang, D. Huang and C. Hu, Nanoscale, 2018, 10, 5997–6004 RSC.
- H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC.
- X. Cheng, R. Sun, L. Yin, Z. Chai, H. Shi and M. Gao, Adv. Mater., 2016, 29, 1604894 CrossRef PubMed.
- Y. Shi, M. Liu, F. Deng, G. Zeng, Q. Wan, X. Zhang and Y. Wei, J. Mater. Chem. B, 2017, 5, 194–206 RSC.
- X. Liu, H. Su, W. Shi, Y. Liu, Y. Sun and D. Ge, Biomaterials, 2018, 167, 177–190 CrossRef CAS PubMed.
- K. Chang, Y. Liu, D. Hu, Q. Qi, D. Gao, Y. Wang, D. Li, X. Zhang, H. Zheng, Z. Sheng and Z. Yuan, ACS Appl. Mater. Interfaces, 2018, 10, 7012–7021 CrossRef CAS PubMed.
- H. Kim, S. Beack, S. Han, M. Shin, T. Lee, Y. Park, S. Kim Ki, K. Yetisen Ali, H. Yun Seok, W. Kwon and K. Hahn Sei, Adv. Mater., 2018, 30, 1701460 CrossRef PubMed.
- J. Meng, X. Chen, Y. Tian, Z. Li and Q. Zheng, Chem. – Eur. J., 2017, 23, 17521–17530 CrossRef CAS PubMed.
- R. Vankayala and C. Hwang Kuo, Adv. Mater., 2018, 30, 1706320 CrossRef PubMed.
- C. Leng, X. Zhang, F. Xu, Y. Yuan, H. Pei, Z. Sun, L. Li and Z. Bao, Small, 2018, 14, 1703077 CrossRef PubMed.
- Z. Liu, Q. Xu, Y. Li and W. Chen, Mater. Chem. Front., 2017, 1, 538–541 RSC.
- Q. Jin, J. Liu, W. Zhu, Z. Dong, Z. Liu and L. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 332–340 CrossRef CAS PubMed.
- Y. Jiang, D. Cui, Y. Fang, X. Zhen, P. K. Upputuri, M. Pramanik, D. Ding and K. Pu, Biomaterials, 2017, 145, 168–177 CrossRef CAS PubMed.
- D.-D. Li, J.-X. Wang, Y. Ma, H.-S. Qian, D. Wang, L. Wang, G. Zhang, L. Qiu, Y.-C. Wang and X.-Z. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 19312–19320 CrossRef CAS PubMed.
- S. Li, X. Wang, R. Hu, H. Chen, M. Li, J. Wang, Y. Wang, L. Liu, F. Lv, X.-J. Liang and S. Wang, Chem. Mater., 2016, 28, 8669–8675 CrossRef CAS.
- J. Zhang, C. Yang, R. Zhang, R. Chen, Z. Zhang, W. Zhang, S.-H. Peng, X. Chen, G. Liu, C.-S. Hsu and C.-S. Lee, Adv. Funct. Mater., 2017, 27, 1605094 CrossRef PubMed.
- T. Sun, J.-H. Dou, S. Liu, X. Wang, X. Zheng, Y. Wang, J. Pei and Z. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 7919–7926 CrossRef CAS PubMed.
- T. Dong, K. Wen, J. Chen, J. Xie, W. Fan, H. Ma, L. Yang, X. Wu, F. Xu, A. Peng and H. Huang, Adv. Funct. Mater., 2018, 28, 1800135 CrossRef.
- Z. Cao, L. Feng, G. Zhang, J. Wang, S. Shen, D. Li and X. Yang, Biomaterials, 2018, 155, 103–111 CrossRef CAS PubMed.
- B. Guo, G. Feng, N. Manghnani Purnima, X. Cai, J. Liu, W. Wu, S. Xu, X. Cheng, C. Teh and B. Liu, Small, 2016, 12, 6243–6254 CrossRef CAS PubMed.
- C. Xie, X. Zhen, Q. Lei, R. Ni and K. Pu, Adv. Funct. Mater., 2017, 27, 1605397 CrossRef.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CAS.
- J. Li, X. Zhen, Y. Lyu, Y. Jiang, J. Huang and K. Pu, ACS Nano, 2018, 12, 8520–8530 CrossRef CAS PubMed.
- Y. Lyu, D. Cui, H. Sun, Y. Miao, H. Duan and K. Pu, Angew. Chem., Int. Ed., 2017, 56, 9155–9159 CrossRef CAS PubMed.
- Q. Miao and K. Pu, Adv. Mater., 2018, 30, 1801778 CrossRef PubMed.
- Y. Jiang and K. Pu, Acc. Chem. Res., 2018, 51, 1840–1849 CrossRef CAS PubMed.
- X. Zhen, C. Xie, Y. Jiang, X. Ai, B. Xing and K. Pu, Nano Lett., 2018, 18, 1498–1505 CrossRef CAS PubMed.
-
S. K. Calderwood, Tumor Ablation, Springer, Netherlands, Dordrecht, 2013, pp. 29–37 Search PubMed.
- M. Koishi, S. I. Yokota, T. Mae, Y. Nishimura, S. Kanamori, N. Horii, K. Shibuya, K. Sasai and M. Hiraoka, Clin. Cancer Res., 2001, 7, 215–219 CAS.
- S.-i. Yokota, M. Kitahara and K. Nagata, Cancer Res., 2000, 60, 2942–2948 CAS.
- D. Yoo, H. Jeong, S.-H. Noh, J.-H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2013, 52, 13047–13051 CrossRef CAS PubMed.
- J. Zhou, M. Li, Y. Hou, Z. Luo, Q. Chen, H. Cao, R. Huo, C. Xue, L. Sutrisno, L. Hao, Y. Cao, H. Ran, L. Lu, K. Li and K. Cai, ACS Nano, 2018, 12, 2858–2872 CrossRef CAS PubMed.
- J. W. Fisher, S. Sarkar, C. F. Buchanan, C. S. Szot, J. Whitney, H. C. Hatcher, S. V. Torti, C. G. Rylander and M. N. Rylander, Cancer Res., 2010, 70, 9855–9864 CrossRef CAS PubMed.
- D. R. Ciocca, N. Cayado-Gutierrez, M. Maccioni and F. D. Cuello-Carrion, Curr. Mol. Med., 2012, 12, 1183–1197 CrossRef CAS PubMed.
- M. Rybiński, Z. Szymańska, S. Lasota and A. Gambin, J. R. Soc., Interface, 2013, 10, 20130527 CrossRef PubMed.
- S. W. Lee, J. W. Lee, J. H. Chung and J. K. Jo, World J. Mens Health, 2013, 31, 247–253 CrossRef PubMed.
- W. Xu and L. Neckers, Clin. Cancer Res., 2007, 13, 1625–1629 CrossRef CAS PubMed.
- T.-y. Lin, W. Guo, Q. Long, A. Ma, Q. Liu, H. Zhang, Y. Huang, S. Chandrasekaran, C. Pan, K. S. Lam and Y. Li, Theranostics, 2016, 6, 1324–1335 CrossRef CAS PubMed.
- W. Guo, P. Reigan, D. Siegel, J. Zirrolli, D. Gustafson and D. Ross, Cancer Res., 2005, 65, 10006–10015 CrossRef CAS PubMed.
- A. Kamal, L. Thao, J. Sensintaffar, L. Zhang, M. F. Boehm, L. C. Fritz and F. J. Burrows, Nature, 2003, 425, 407 CrossRef CAS PubMed.
- J. Davenport, J. R. Manjarrez, L. Peterson, B. Krumm, B. S. J. Blagg and R. L. Matts, J. Nat. Prod., 2011, 74, 1085–1092 CrossRef CAS PubMed.
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed.
- X. Chen, Z. Zhang, Z. Ding, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 10376–10380 CrossRef CAS PubMed.
- C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2017, 60, 450–459 CrossRef CAS.
- R. Zhao, C. Dou, J. Liu and L. Wang, Chin. J. Polym. Sci., 2017, 35, 198–206 CrossRef CAS.
- T. Sun, J. Qi, M. Zheng, Z. Xie, Z. Wang and X. Jing, Colloids Surf., B, 2015, 136, 201–206 CrossRef CAS PubMed.
- W. Wang, L. Wang, Y. Li, S. Liu, Z. Xie and X. Jing, Adv. Mater., 2016, 28, 9320–9325 CrossRef CAS PubMed.
- Y. Li, C. Jiang, D. Zhang, Y. Wang, X. Ren, K. Ai, X. Chen and L. Lu, Acta Biomater., 2017, 47, 124–134 CrossRef CAS PubMed.
- Y. Zhang, Z. Yang, X. Tan, X. Tang and Z. Yang, AAPS PharmSciTech, 2017, 18, 1987–1997 CrossRef CAS PubMed.
- Y. Yang, L. Yang, Q.-D. You, F.-F. Nie, H.-Y. Gu, L. Zhao, X.-T. Wang and Q.-L. Guo, Cancer Lett., 2007, 256, 259–266 CrossRef CAS PubMed.
- X. Zhu, H. Zhang, Y. Lin, P. Chen, J. Min, Z. Wang, W. Xiao and B. A. Chen, J. Chemother., 2009, 21, 666–672 CrossRef CAS PubMed.
- M. K. Pandey, B. Sung, K. S. Ahn, A. B. Kunnumakkara, M. M. Chaturvedi and B. B. Aggarwal, Blood, 2007, 110, 3517–3525 CrossRef CAS PubMed.
- J. Chen, H.-Y. Gu, N. Lu, Y. Yang, W. Liu, Q. Qi, J.-J. Rong, X.-T. Wang, Q.-D. You and Q.-L. Guo, Life Sci., 2008, 83, 103–109 CrossRef CAS PubMed.
- L. Zhao, Q.-L. Guo, Q.-D. You, Z.-Q. Wu and H.-Y. Gu, Biol. Pharm. Bull., 2004, 27, 998–1003 CrossRef CAS.
- J. Wu, L. Zhao, X. Xu, N. Bertrand, W. Choi II, B. Yameen, J. Shi, V. Shah, M. Mulvale, L. MacLean James and C. Farokhzad Omid, Angew. Chem., Int. Ed., 2015, 54, 9218–9223 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization, synthesis and additional figures. See DOI: 10.1039/c8qm00459e |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Xingxing
Chen
ab,
Xin
Wang
c,
Shi
Liu
a,
Jun
Liu
ab,
Xingxing
Chen
ab,
Xin
Wang
c,
Shi
Liu
a,
Jun
Liu