
S,N co-doped reduced graphene oxide sheets with cobalt hydroxide nanocrystals for highly active and stable bifunctional oxygen catalysts†
 *a
*a
Abstract
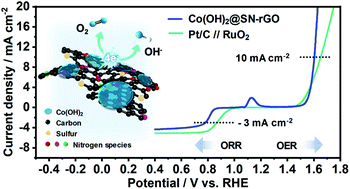
It is imperative to develop cost-effective and stable cathode materials with satisfactory activities for both oxygen evolution and oxygen reduction reactions (OER and ORR) in order to build next-generation rechargeable metal–air battery systems. Herein, we developed an effective strategy for fabricating highly active and stable bifunctional oxygen catalysts using a highly active ORR catalyst and hybridizing it with an OER catalyst. The effects of single (S)- and multi (S and N)-doping on the ORR catalytic efficiency of reduced graphene oxide (rGO) sheets were investigated. Cobalt hydroxide (Co(OH)2) nanocrystals with diameters of less than 5 nm were directly grown on SN-rGO via a simple precipitation method at room temperature to produce a bifunctional oxygen catalyst (Co(OH)2@SN-rGO). The Co(OH)2@SN-rGO bifunctional oxygen catalyst exhibited outstanding catalytic activities for both OER and ORR and showed excellent stability up to 40 h. The results demonstrated that the bifunctional oxygen electrode activities of the non-noble metal-based catalyst prepared in this study were as high as those of commercial noble metal-based oxygen electrodes.
 Please wait while we load your content...
Please wait while we load your content...

