
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical properties of chromium oxyfluoride CrO2−xFx with 0 ≤ x ≤ 0.3†
Kazuhiko
Mukai
 *a,
Takeshi
Uyama
a and
Ikuya
Yamada
b
*a,
Takeshi
Uyama
a and
Ikuya
Yamada
b
aToyota Central Research & Development Laboratories, Inc., Yokomichi 41–1, Nagakute, Aichi 480–1192, Japan. E-mail: e1089@mosk.tytlabs.co.jp; Fax: +81-561-63-6119; Tel: +81-561-71-7698
bDepartment of Materials Science, Graduate School of Engineering, Osaka Prefecture University, 1–2 Gakuen, Sakai, Osaka 599–8570, Japan
First published on 26th September 2019
Abstract
To overcome the limitations of graphite as a negative electrode material for lithium-ion batteries (LIBs), transition metal oxyfluorides are under active development. In this study, chromium oxyfluorides CrO2−xFx with 0 ≤ x ≤ 0.3 were synthesized under a high-pressure/high-temperature (HP/HT) environment, and their electrochemical properties were examined in a nonaqueous lithium cell. The HP/HT-treated CrO2 maintained a rutile structure and exhibited a rechargeable capacity (Qrecha) of over 400 mA h g−1 at 298 K. The replacement of O2− ions with F− ions in CrO2 was confirmed by linear changes in the tetragonal lattice parameters, weaker ferromagnetic interactions between Cr4+ ions, and elemental mappings of F− ions. The Qrecha values of the x > 0 samples at 298 K decreased to 150–300 mA h g−1 because of low electric conductivity in CrO2−xFx. However, the Qrecha values at 318 K increased to 600–700 mA h g−1, and the cycle performance over 30 cycles was better than that of the HP/HT-treated CrO2 sample with no F− substitution. Hence, CrO2−xFx was found to be a promising negative electrode material for LIBs, although its cycle stability should be further improved.
1. Introduction
Transition metal (M) oxyfluorides with the general formula MaObFc have recently received a great deal of attention as an electrode material for lithium-ion batteries (LIBs), although their synthesis and characterization date back to the 1950s.1–3 The relevant electrochemical reaction is represented by| MaObFc + (2b + c)Li+ + (2b + c)e− ↔ aM + bLi2O + cLiF, | (1) |
Table 1 summarizes the crystal structures and electrochemical properties of the MaObFc compounds reported thus far.6–22 NbO2F,6,7 TiF27,8,9,10 TaO2F,11 and VO2F12,13,14 have a regular or distorted ReO3-type structure with a rechargeable capacity (Qrecha) of 200–400 mA h g−1. Overall, their operating voltage as a function of Qrecha is featureless, suggesting the same conversion reaction that is typical for their parent MO compounds.23,24 On the other hand, FeOxF2−x with a rutile structure15–19 and BiOxF3−2x with a tysonite-like structure20,21 exhibited a flat operating voltage at ∼2.4 and ∼3.0 V, respectively, indicating a two-phase reaction. The Qrecha values of FeOxF2−x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15–18 and BiOxF3−2x20,21 are ∼400 and ∼200 mA h g−1, respectively. An amorphous-like MnOFx phase22 first showed a flat operating voltage at ∼4.5 V owing to the release of oxygen from the lattice and then indicated a Qrecha of ∼225 mA h g−1 at 0.0–3.0 V.
15–18 and BiOxF3−2x20,21 are ∼400 and ∼200 mA h g−1, respectively. An amorphous-like MnOFx phase22 first showed a flat operating voltage at ∼4.5 V owing to the release of oxygen from the lattice and then indicated a Qrecha of ∼225 mA h g−1 at 0.0–3.0 V.
| Compound | Crystal structure | Space group | Electrochemical properties | Synthesis method | Ref. |
|---|---|---|---|---|---|
| NbO2F | ReO3-type (cubic) |
Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
200 mA h g−1 (0.005–3.0 V) | Solution route | 7 |
| TiOF2 | (Distorted) ReO3-type (cubic or rhombohedral) |
Pmm or Rc |
400 mA h g−1 (0.005–3.0 V) | Solution route | 7 |
| TaO2F | ReO3-type (cubic) |
Pmm |
Unknown | Solution route | 11 |
| VO2F | Distorted ReO3-type (rhombohedral) |
Rc |
200 mA h g−1 (2.0–4.5 V) | Ball-milling or HP | 14 |
| FeOxF2−x | Rutile (tetragonal) | P42/mnm | 450 mA h g−1 (1.6–4.4 V) | Solid-state reaction | 15 |
| BiOxF3−2x | Tysonite-like (orthorhombic) | Pnma | 180 mA h g−1 (2.0–4.5 V) | Solution route | 21 |
| MnOFx | Amorphous | 225 mA h g−1 (1.5–4.5 V) | Ball milling | 22 | |
| CrO2−xFx | Rutile (tetragonal) | P42/mnm | 100–400 mA h g−1 (0.02–3.0 V) | HP | This work |
As understood by Table 1, all the MaObFc compounds, except for TiOF2, indicated higher operating voltages above 1.5 V or lower Qrecha values below 300 mA h g−1. Since the operating voltage and Qrecha of graphite are ∼0.2 V and ∼330 mA h g−1, respectively,4,5 the energy density of LIBs would be decreased when employing the MaObFc compounds as a negative electrode. Moreover, MaObFc compounds are usually synthesized via solution routes using highly toxic, corrosive fluorine-containing acids or by ball milling a mixture of transition metal oxides and LiF.6–22 These methods provide a bulky electrode material, lowering the volumetric energy density of LIBs. Hence, a denser MaObFc compound with a Qrecha > 330 mA h g−1 below ∼1.0 V is required, to surpass the electrochemical properties of graphite and further increase the energy density of LIBs.4,5
In this study, we synthesized a series of CrO2−xFx with 0 ≤ x ≤ 0.3 using a high-pressure/high-temperature (HP/HT) method, and we examined their electrochemical properties, because chromium oxides, such as Cr2O325–27 and Cr3O8,25,28 have shown operating voltages below 1.0 V with ∼500 mA h g−1. Furthermore, O2− ions in CrO2 have easily been substituted with F− ions under HP environments above 6 GPa,2,3 providing dense CrO2−xFx compounds. Here, CrO2−xFx, which has a rutile (tetragonal) structure with the P42/mnm space group, exhibits ferromagnetic behavior above room temperature and is regarded as a promising spintronic material because of its nearly 100% spin polarization at the Fermi level.3,29–32 To the best of our knowledge, the electrochemical properties of CrO2−xFx including CrF2 have never been reported, except for amorphous CrO2−x compounds with 0 ≤ x ≤ 0.5.33 Here, the CrO2−xFx samples demonstrated a Qrecha of 100–400 mA h g−1 at 298 K and a Qrecha of 600–700 mA h g−1 at 318 K. The structural, magnetic, and electrochemical properties as a function of x in CrO2−xFx are discussed in detail.
2. Experimental
2.1. Sample preparation
Polycrystalline CrO2 and CrF2 powders provided by Kojundo Chemical Laboratory Co., Ltd (Japan) were mixed in various ratios using a mortar and a pestle in an argon-filled glove-box and then separately pressed into pellets with a diameter of 2.8 mm and a height of ∼4 mm. The molar ratios of the different CrO2/CrF2 mixtures were 9:1, 8:2, 7:3, 6:4, or 4:6, forming a general chemical formula of CrO2−xFx with x = 0.05, 0.1, 0.15, 0.2, or 0.3. After being packed into a (Mg,Co)O pressure medium (Mino Ceramics Co., Ltd), the pellet was heated at 1273 K for 30 min under 12 GPa. The pellet consisting of only CrO2 powder was also treated under the same conditions, and this sample is denoted by HP/HT-treated CrO2. The HP/HT synthesis and experimental setup were described in detail elsewhere.34–37
2.2. Characterization
The particle morphologies of the synthesized samples were investigated using a scanning electron microscope (SEM; S-3600 N, Hitachi High-Technologies Co., Ltd). Approximately 1 mg of sample, which was attached onto the sample holder with carbon tape, was coated with electrically conducting Au particles (IB-3, Eiko Co., Ltd). To clarify the temperature at which the rutile structure converts to corundum, thermogravimetric/differential thermal analysis (TG/DTA) was conducted on pristine CrO2 powder (Thermo plus EVO2, TG-DTA 8122, Rigaku Co., Ltd). Specifically, the sample was heated to 1273 K at 5 K min−1 under air flowing at 100 mL min−1.X-ray diffraction (XRD) measurements were performed at the BL5S2 beamline of the Aichi Synchrotron radiation center. Each sample was inserted into a glass capillary tube with a diameter of 0.3 mm (WHM-Glas Müller Bmbh) to eliminate the effects of selective orientation. XRD patterns of the samples were recorded in the 2θ range between 5 and 90° using Pilatus 100 K detectors. The X-ray wavelength was determined to be 0.799420(3) Å using a silicon standard (NIST 640d). Rietveld analyses were conducted using RIEATN-FP software,38 and crystal structures were drawn using VESTA software.39
The magnetic susceptibilities (χ) were examined using a superconducting quantum interference device (SQUID) magnetometer (MPMS, Quantum Design). χ was recorded in field-cooling (FC) mode with a magnetic field (H) of 10 kOe while the temperature decreased from 400 K to 5 K. Magnetization (M) vs. H curves were also recorded at 5 K in the H range between −55 and 55 kOe.
For only the x = 0.2 sample, the distribution of Cr, O, and F atoms in the particles were examined using an electron probe microanalyzer (EPMA; JXA-8500F, JEOL Ltd) with an accelerating voltage of 15 kV. This is because as described later, the particle size of CrO2−xFx drastically changed at x ≥ 0.2. The sample was embedded in epoxy resin and then cut into a rectangle form using a cross-sectional polisher (SM-09010, JEOL Ltd) equipped with Ar+ ions. The experimental procedure was similar to the one we used for our recent EPMA analysis of LiCo0.64Mn0.36O2.37
2.3. Electrochemical measurements
Discharge and charge profiles were recorded using a nonaqueous lithium cell. The electrolyte was 1 M LiPF6 dissolved in ethylene carbonate (EC)/diethylene carbonate (DEC) (EC/DEC = 1/1 by volume, Kishida Chemical Co., Ltd). A mixed electrode consisting of 70 wt% active material, 20 wt% conducting carbon (acetylene black, HS-100, Denka Co., Ltd), and 10 wt% polytetrafluoroethylene (PTFE) was used as the working electrode (diameter = 10 mm), while lithium metal pressed onto a stainless steel plate (diameter = 19 mm) was used as the counter electrode. After being fabricating in the argon-filled glove-box, the lithium cells were operated at a current of 0.1 or 0.5 mA (≃0.1 or 0.5 mA cm−2) between 0.02 and 3.0 V. The cells were tested at both 298 and 318 K. The lithium cells with the x = 0 and 0.2 samples were cycled over 30 times at 298 and 318 K, and the rest of lithium cells were cycled over 5 cycles at 298 K. Above cycle tests were performed with a current of 0.1 mA. In addition, the lithium cells with x = 0.05, 0.15, 0.2, and 0.3 were cycled over 50 cycles at 318 K with a current of 0.5 mA.To clarify the reaction mechanism, ex situ XRD was performed at the BL5S2 beamline of the Aichi Synchrotron radiation center. The lithium cells with the x = 0 and 0.2 samples were discharged down to 0.02 V, and then, in the argon-filled glove-box, the electrodes were removed. Each sample was packed into a glass capillary tube with a diameter of 0.7 mm (WHM-Glas Müller Bmbh), and XRD data were then recorded as for the initial samples. XRD patterns were also taken after the 30-cycle test (x = 0 and 0.2). The X-ray wavelength was determined to be 0.799436(2) Å. The experimental conditions are described in further detail in the results and discussion section.
3. Results and discussion
3.1. HP/HT-treated CrO2
Fig. 1a and b show SEM images of pristine CrO2 and HP/HT-treated CrO2 samples. Particles of the pristine CrO2 exhibit a flake-like morphology with a lateral size of 5–50 μm and a thickness of ∼2 μm. These flake-like particles consisted of numerous nanoscale particles, as shown in Fig. S1.† Particles of HP/HT-treated CrO2 exhibit a non-uniform morphology with a smooth surface. The average size of the primary particles, which are aggregates of pristine CrO2 particles, is approximately 20 μm.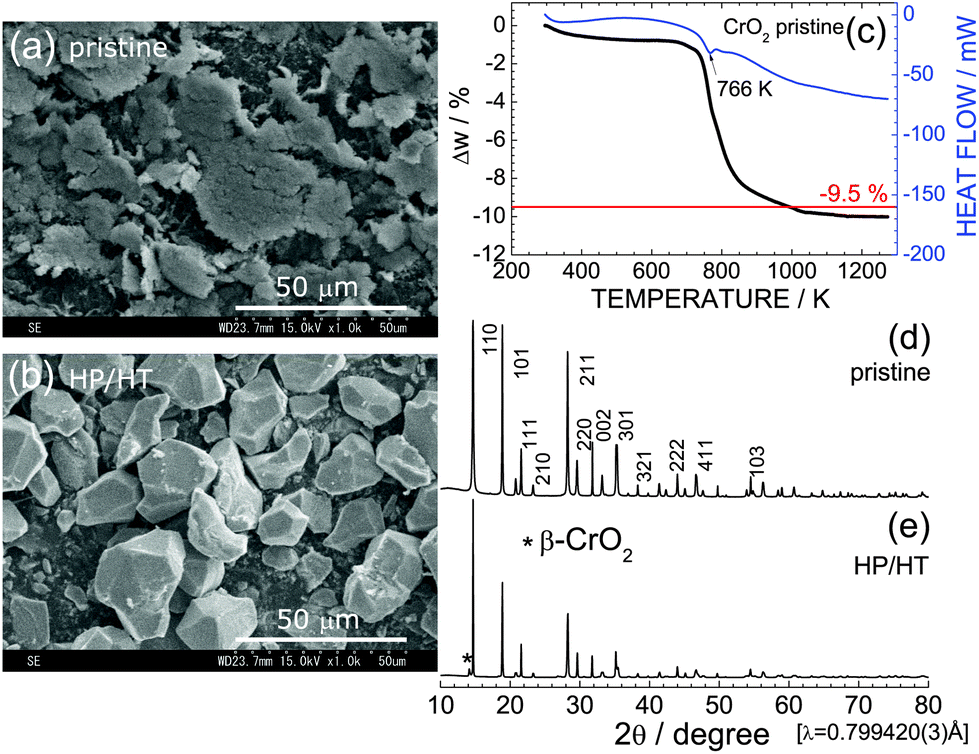 | ||
| Fig. 1 SEM images of the (a) pristine CrO2 and (b) HP/HT-treated CrO2 samples. (c) The TG/DTA curve of the pristine CrO2 sample. Synchrotron XRD patterns of the (d) pristine CrO2 and (e) HP/HT-treated CrO2 samples. The * symbol indicates the diffraction line from the β-CrO2 phase. | ||
Fig. 1c shows the TG/DTA curve of the pristine CrO2 sample to clarify its thermal stability under ambient pressure. The weight of CrO2 rapidly decreases starting at 766 K, and levels off to an almost constant value above 1073 K. The change in weight (Δw) above 1073 K was ∼−10.0%, which was consistent with the calculated Δw value (=−9.5%) based on the transformation of CrO2 with a rutile structure into Cr2O3 with a corundum structure:
| 2CrO2 → Cr2O3 + 1/2O2↑. | (2) |
Despite the oxygen loss under ambient pressure, the HP environment stabilized the crystal structure of CrO2. Fig. 1d and e show XRD patterns of the pristine CrO2 and HP/HT-treated CrO2 samples, respectively. The pristine CrO2 sample contains only the rutile phase with lattice parameters of at = 4.4224(1) Å and ct = 2.9160(1) Å. The corresponding Rietveld analysis results are shown in Fig. S2a.† The crystal structure of the HP/HT-treated CrO2 sample is also assigned to the rutile structure, although a small amount of β-CrO2 phase coexists in the sample (indicated by * in Fig. 1e). β-CrO2, which considered a HP-form of CrO2, adopts a distorted rutile structure with the Pnnm space group.29 According to the Rietveld analysis results shown in Fig. S2b,† the weight fraction of the β-CrO2 phase was estimated to be 6.5%. The lattice parameters of the major CrO2 phase were determined to be at = 4.4182(2) Å and ct = 2.9199(1) Å, which were comparable to those of the pristine CrO2 sample.
Fig. 2a and b show the discharge and charge curves of the pristine CrO2 and HP/HT-treated CrO2 samples, respectively, operating at 298 K. Each discharge curve for the first cycle was discarded to ignore the contributions of the decomposition reaction of the PTFE binder. The voltage of the pristine CrO2 rapidly drops to ∼0.4 V at the beginning of the discharge reaction and then maintains a constant value at ∼0.2 V. The discharge capacity (Qdis) reaches 377 mA h g−1. The subsequent charge curve is quite different from the discharge curve, indicating a large voltage hysteresis of ∼0.8 V. This is a typical characteristic of the conversion reaction, which occurs in various MaObFc6–22 and MO23,24 compounds.
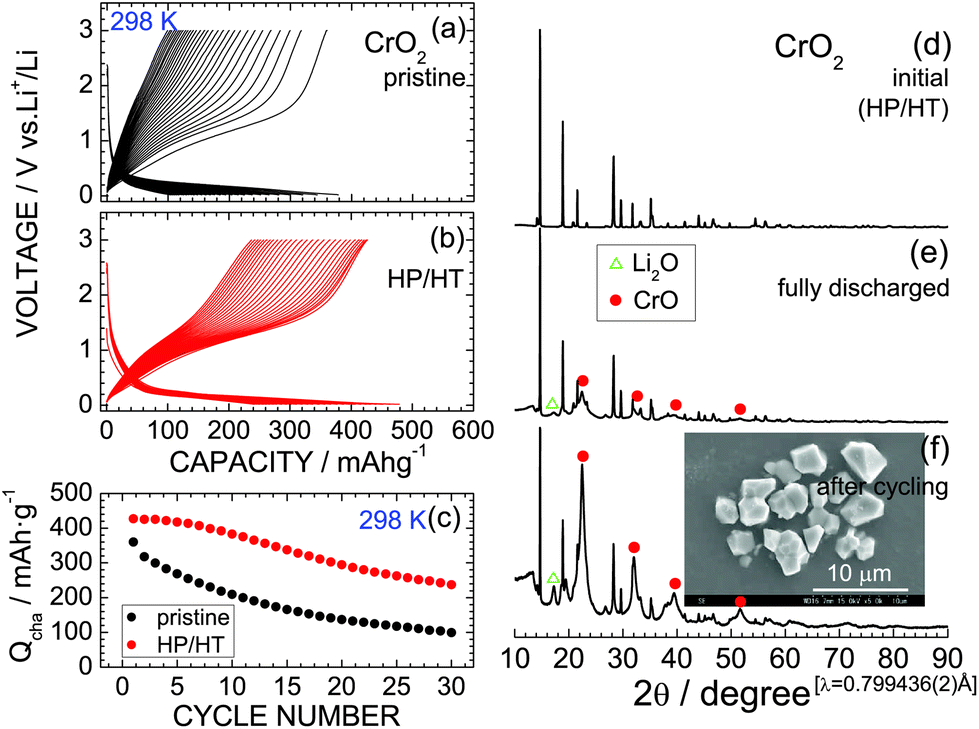 | ||
| Fig. 2 Discharge and charge curves of the (a) pristine CrO2 and (b) HP/HT-treated CrO2 samples operated at 0.1 mA and 298 K. (c) Cycling performance of the two CrO2 samples at 298 K. Ex situ XRD patterns of the HP/HT-treated CrO2 samples: (d) initial state, (e) fully discharged state at 0.1 V, and (f) after cycling over 30 times. Inset: SEM image of the sample after cycling. | ||
The discharge and charge curves of the HP/HT-treated CrO2 sample were similar to those for the pristine CrO2 sample; however, the values of Qdis and charge capacity (Qcha) differed. Specifically, the maximum Qcha values for the pristine and HP/HT-treated CrO2 samples were 360.1 and 427.0 mA h g−1, respectively. Moreover, as shown in Fig. 2c, the cycling performance of the HP/HT-treated CrO2 was superior to that of the pristine CrO2. In addition, the pristine CrO2 retained 27.6% of its capacity over 30 cycles, and the HP/HT-treated CrO2 retained 55.6%. Note that this cycling test lasted for over one month because of the low current density of 0.1 mA cm−2. This is a severe electrochemical test as the cells were exposed to low voltages below 0.5 V for a long time. The cycle test at such low current densities has also been applied to the preliminary evaluation of LiNi1/2Mn1/2O240 and LiCo1/3Ni1/3Mn1/3O2.41
The complete conversion reaction is represented by
| CrO2 + 4Li+ + 4e− ↔ Cr + 2Li2O, | (3) |
m space group (PDF-4+01-073-9520).45 The cubic lattice parameter (ac) of the CrO phase was calculated to be ∼4.10 Å. The diffraction line at 2θ = 17.3° (d ≃ 2.66 Å) is indexed to the 111 diffraction line of Li2O (PDF-4+04-008-3420).45 Continuous cycling strengthens the intensities of the CrO and Li2O phase peaks, thus demonstrating that the initial CrO2 phase transforms into an amorphous-like phase with continued cycling. The actual electrochemical reaction of CrO2 is thus represented by| CrO2 + 2Li+ + 2e− ↔ CrO + Li2O, | (4) |
3.2. CrO2−xFx
Fig. 3a–e show the XRD patterns of the CrO2−xFx samples with x = 0.05, 0.1, 0.15, 0.2, and 0.3. All the XRD patterns were assigned to the rutile structure, similar to the x = 0 samples. The Cr2O3 impurity was observed in the x = 0.2 and 0.3 samples. The at and ct values, which were determined via Rietveld analyses, are shown in Fig. 3f and g, respectively, which also show reported at and ct values.3 The lattice parameters at and ct in this study increase almost monotonically with x, indicating that as expected, O2− ions are substituted by F− ions. The lattice volume (V) obtained by at2 × ct are shown in Fig. S3.† However, these lattice parameters were slightly lower than those of the reported values,3 particularly at x ≥ 0.2. Recently, anion ordering between O2− and F− ions appeared in the electron diffraction patterns of CrO2−xFx with x = 0.1, 0.12, and 0.14.31 In contrast, because the X-ray scattering factors of O2− and F− ions are similar to each other in this study, such anion ordering is not observed in the XRD measurements. | ||
| Fig. 3 Synchrotron XRD patterns of the CrO2−xFx samples with (a) x = 0.05, (b) x = 0.1, (c) x = 0.15, (d) x = 0.2, and (e) x = 0.3. An SEM image of each sample is shown in the inset. Diffraction lines from the Cr2O3 phase are indicated by circles. Lattice parameters (f) at and (g) ct as a function of x in CrO2−xFx, including the at and ct values reported in ref. 2. | ||
The SEM images in the inset of Fig. 3a–e clearly show that substituting F− for O2− altered the particle sizes of CrO2−xFx. The average size of primary particles is approximately 10 μm for the x ≤ 0.15 samples, whereas it is less than 3 μm for the x = 0.2 and 0.3 samples. The drastic change in the particle size was also observed on the HP/HT studies on Li[Li1/3Ti5/3]O4.34 Enlarged SEM images for the x = 0.2 and 0.3 samples are shown in Fig. S4.†
Since CrO2 exhibited ferromagnetic behavior with a Curie temperature (TC) of ∼390 K,3,29–32 the effects of substituting F− for O2− ions were investigated from the aspect of magnetism. Fig. 4a and b show the temperature dependence of χ and χ−1 measured in the FC mode with H = 10 kOe. As the temperature decreases from 400 K, χ of the pristine CrO2 sample increases from ∼0.5 emu mol−1 to ∼0.9 emu mol−1 and then maintains an almost constant value (∼1.0 emu mol−1) below 200 K. Similar behavior is also observed in the χ (or χ−1) vs. temperature curves of the x = 0, 0.05, 0.1, 0.15, and 0.2 samples. However, the x = 0.3 sample indicates a slightly different trend, especially below 100 K, wherein χ rapidly drops to ∼0.45 emu mol−1 at 5 K. This decrease in χ is attributed to partial antiferromagnetic Cr4+–Cr3+ interactions produced by the F− ion substitution.29,30 Antiferromagnetic interactions also appear in the M–H curves at 5 K (Fig. 3c). The pristine CrO2 sample shows soft ferromagnetic behavior with a maximum (saturation) M (Mmax) of ∼11900 emu mol−1. This Mmax value corresponds to ∼2.1μB, which is consistent with the spin-only calculated magnetic moment (=2μB) assuming that Cr4+ ions are in the t2g2 (S = 1) state. Substituting F− ions for O2− ions decreases the Mmax value, except for the x = 0.2 sample (Fig. 4d). Moreover, the M–H curve for x = 0.3 is a competitive consequence of ferromagnetic interactions between Cr4+ ions and antiferromagnetic interactions between Cr4+ and Cr3+ ions.
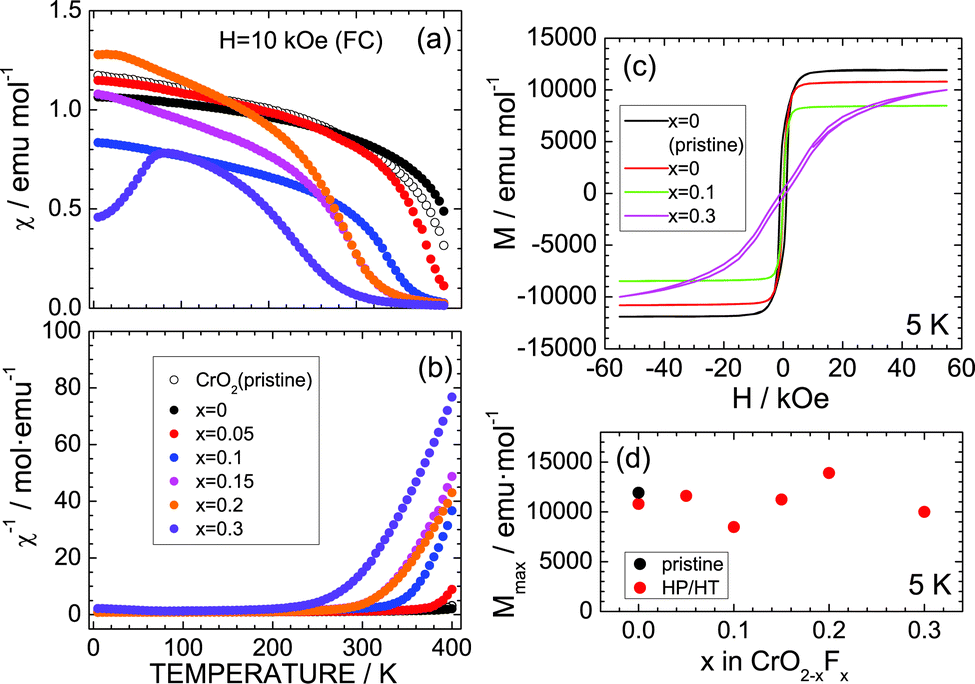 | ||
| Fig. 4 (a) χ and (b) χ−1 of the CrO2−xFx samples with x = 0, 0.05, 0.1, 0.15, 0.2, and 0.3. (c) M–H curve of the CrO2−xFx samples with x = 0, 0.1, and 0.3 at 5 K. The two x = 0 samples in (a)–(c) are the pristine CrO2 and HP/HT-treated CrO2 samples. (d) M at H = 55 kOe, i.e., Mmax, as a function of x in CrO2−xFx. | ||
To clarify the distributions of O and F atoms in CrO2−xFx particles, Fig. 5 shows the EPMA mapping results from the x = 0.2 sample for O and F atoms. O and F atoms are distributed throughout the entire particles, but F is slightly localized near the surface of the particles. This means that the F− ions are inhomogeneously distributed inside the particles, although the lattice parameters at and ct increased almost linearly with x. Cr, O, and F atoms are mapped at the 1 μm scale in Fig. S5.†
 | ||
| Fig. 5 EPMA results from the x = 0.2 sample: mappings of (a) O and (b) F and (c) overlapping mapping of O and F atoms. (d) Corresponding SEM image. Scale bars: 2 μm scale. | ||
Fig. 6a–e show the discharge and charge curves of the x = 0.05, x = 0.1, x = 0.15, x = 0.2, and x = 0.3 samples operated at 298 K. For comparison, the discharge and charge curves of CrF2 are also shown in Fig. 6f. For the x = 0.05 sample, the Qdis and Qcha values during the initial cycle are 399.0 and 364.4 mA h g−1, respectively, which are slightly lower than those for the HP/HT-treated CrO2 (x = 0) sample. However, as x increases from x = 0.05, both Qdis and Qcha decrease to 150–300 mA h g−1 due to a low electrical conductivity, which has also been observed with other oxyfluoride materials.6–22 Since the operating voltage of the discharge reaction was close to the discharge cut-off voltage ( = 0.02 V), the discharge reaction of the CrO2−xFx compounds finished before storing a large amount of Li ions. Note that CrF2 exhibits a Qrecha of ∼100 mA h g−1 and a capacitor-like voltage profile.
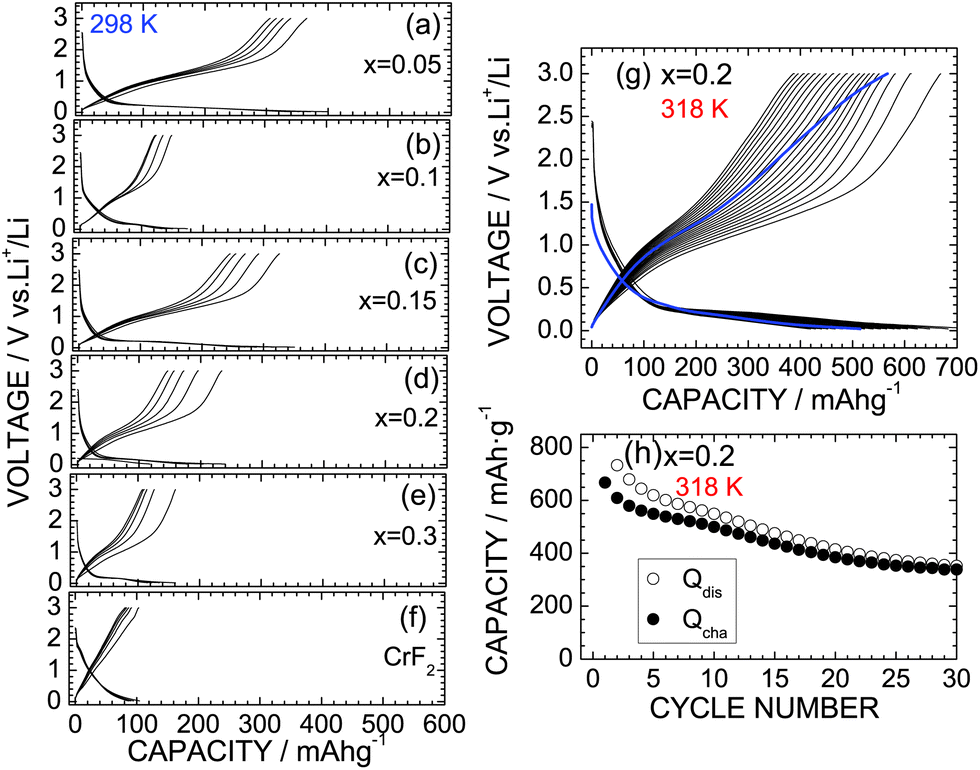 | ||
| Fig. 6 Discharge and charge curves of the CrO2−xFx samples with (a) x = 0.05, (b) x = 0.1, (c) x = 0.15, (d) x = 0.2, (e) x = 0.3, and (f) CrF2 operated at 298 K. (g) Discharge and charge curves of the x = 0.2 sample operated at 318 K. (h) Cycling performance of the x = 0.2 sample at 318 K. The blue lines in (c) indicate the discharge and charge curves at a low current density of ∼0.03 mA cm−2 after the cycle test. | ||
The lithium cells with the x = 0 and 0.2 samples were also cycled at 318 K to clarify the cycle stability at high temperatures. Fig. 6g shows the discharge and charge curves of the x = 0.2 sample at 318 K with a current of 0.1 mA, and Fig. 6h shows the corresponding cycle performance over 30 cycles. The Qdis and Qcha values during the initial cycle are 773.1 and 667.3 mA h g−1, respectively, which are approximately three times larger than those at 298 K. Although the Qcha value decreases with cycling, it remains at 338.8 mA h g−1 after 30 cycles, thus providing a capacity retention of 50.8%. Note that this capacity retention is superior to that of the HP/HT-treated CrO2 at 318 K, wherein 40.0% of the capacity was retained (see Fig. S6†). Furthermore, as shown in Fig. S7,† the capacity fading at 318 K was suppressed by the substitution of F ions, especially at the latter of 50 cycles. According to the ex situ XRD measurements, the CrO phase with ac = 4.09 Å was produced in the fully discharged state (Fig. S8b†). Furthermore, the diffraction line at 2θ ≃ 17.3° was hardly observed, indicating that the substitution of F− ions suppresses the formation of the Li2O phase. This is probably due to the strong ionic interaction between the Cr4+ and F− ions. Since the initial rutile structure of CrO2−xFx transformed into an amorphous phase after the cycling test (Fig. S8c†), the discharge and charge reactions of CrO2−xFx proceed between the CrO and amorphous phases, unlike the electrochemical reaction described in eqn (1).
Finally, we discuss the significance of the CrO2−xFx materials as a negative electrode for LIBs. The operating voltage in the discharge reaction was close to 0.2 V, which is the lowest operating voltage observed for oxyfluorides (Table 1) and is similar to that of the graphite. Furthermore, the theoretical density of CrO2−xFx according to the XRD data is ∼4.89 g cm−3, which is two times larger than that of the graphite (≃2.2 g cm−3). Since LIBs are the device that functions in a limited space, CrO2−xFx is a promising negative electrode material for LIBs in terms of volumetric energy density. However, even with the x = 0.2 sample, the cycle stability of CrO2−xFx is not sufficient for practical LIB applications. According to the electrochemical measurements after the cycle test at 318 K, both Qdis and Qcha values recovered to ∼87% of their initial values at the low current density of ∼0.03 mA cm−2 (Fig. 6g). Hence, the capacity fading in CrO2−xFx is caused not by a destruction of the core material but by a failure of contact between the active material and the conducting carbon. Further optimizing the electrode mixture and the type of conducting carbon and/or binders could improve the cycle performance of CrO2−xFx.
4. Conclusion
The HP/HT-treated CrO2 maintained the rutile structure even after heating at 1273 K and exhibited a Qrecha of 400 mA h g−1 at 298 K. The operating voltage of the discharge reaction was close to 0.2 V, which is the lowest reported operating voltage among oxyfluoride materials such as FeOxF2−x and BiOxF3–2x. F− ions successfully replaced the O2− ions in CrO2 under the HP/HT environment, providing a linear increase in the lattice parameters at and ct and weakening the ferromagnetic interaction between Cr4+ ions. The Qrecha values of the x > 0 samples decreased to 150–300 mA h g−1 at 298 K but increased to more than 600 mA h g−1 at 318 K. The cycle performance of x = 0.2 at 318 K was superior to that of the HP/HT-treated CrO2 sample, suggesting that the substitution of F− ions stabilized the crystal lattice during the discharge and charge reactions at high temperatures. The electrochemical properties of CrO2−xFx are promising as an alternative of graphite, the most popular negative electrode material in LIBs. Furthermore, the present results provide a new direction for synthesizing (lithium) oxyfluoride materials with a large volumetric energy density.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Dr Takao Inoue of TCRDL for help with the HP/HT syntheses and Mr Yusuke Yagi of TCRDL for the EPMA analysis. The synchrotron XRD patterns were measured at the BL5S2 beamline of the Aichi Synchrotron Radiation Center, Aichi Science & Technology Foundation, Japan (Proposal No. 201803038 and 201901008).References
- K. Vorres and J. Donohue, Acta Crystallogr., 1955, 8, 25 CrossRef CAS.
- B. L. Chamberland, A. W. Sleight and W. H. Cloud, J. Solid State Chem., 1970, 2, 49 CrossRef CAS.
- B. L. Chamberland, C. G. Frederick and J. L. Gillson, J. Solid State Chem., 1973, 6, 561 CrossRef CAS.
- T. Ohzuku, Y. Iwakoshi and K. Sawai, J. Electrochem. Soc., 1993, 140, 2490 CrossRef CAS.
- K. Mukai, T. Inoue and M. Hasegawa, J. Power Sources, 2017, 366, 185 CrossRef CAS.
- C. Bohnke, J. L. Fourquet, N. Randrianantoandro, T. Brousse and O. Crosnier, J. Solid State Electrochem., 2001, 5, 1 CrossRef CAS.
- M. V. Reddy, S. Madhavi, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2006, 162, 1312 CrossRef CAS.
- B. Li, D. Wang, Y. Wang, B. Zhu, Z. Gao, Q. Hao, Y. Wang and K. Tang, Electrochim. Acta, 2015, 180, 894 CrossRef CAS.
- C. Evangelisti, M. Hayatifar, F. Marchetti, M. Marelli, G. Pampaloni and F. Piccinelli, Inorg. Chem., 2016, 55, 1816 CrossRef CAS PubMed.
- N. Luovain, Z. Karkar, M. El-Ghozzi, P. Bonnet, K. Guérin and P. Willmann, J. Mater. Chem. A, 2014, 2, 15308 RSC.
- J. Dabachi, M. Body, C. Galven, F. Boucher and C. Legein, Inorg. Chem., 2017, 56, 5219 CrossRef CAS PubMed.
- J. C. Pérez-Flores, R. Villamor, D. Ávila-Brande, J. M. Gallardo Amores, E. Morán, A. Kuhn, F. García-Alvarado, M. Bervas, B. Yakshinskiy, L. C. Klein and G. G. Amatucci, J. Mater. Chem. A, 2015, 3, 20508 RSC.
- R. Chen, E. Maawad, M. Knapp, S. Ren, P. Beran, R. Witter and R. Hempelmann, RSC Adv., 2016, 6, 65112 RSC.
- M. A. Cambaz, B. P. Vinayan, O. Clemens, A. R. Munnangi, V. S. K. Chakravadhanula, C. Kübel and M. Fichtner, Inorg. Chem., 2016, 55, 3789 CrossRef CAS PubMed.
- N. Pereira, F. Badway, M. Wartelsky, S. Gunn and G. G. Amatucci, J. Electrochem. Soc., 2009, 156, A407 CrossRef CAS.
- I. D. Gocheva, I. Tanaka, T. Doi, S. Okada and J. Yamaki, Electrochem. Commun., 2009, 11, 1583 CrossRef CAS.
- L. Li, F. Meng and S. Jin, Nano Lett., 2012, 12, 6030 CrossRef CAS PubMed.
- S.-W. Kim, N. Pereira, N. A. Chernova, F. Omenya, P. Gao, M. S. Whittingham, G. G. Amatucci, D. Su and F. Wang, ACS Nano, 2015, 10, 10076 CrossRef PubMed.
- M. Burbano, M. Duttine, B. J. Morgan, O. J. Borkiewicz, K. W. Chapman, A. Wattiaux, A. Demourgues, H. Groult, M. Salanne and D. Dambournet, J. Phys. Chem. Lett., 2019, 10, 107 CrossRef CAS PubMed.
- M. Bervas, B. Yakshinskiy, L. C. Klein and G. G. Amatucci, J. Am. Ceram. Soc., 2006, 89, 645 CrossRef CAS.
- M. Bervas, L. C. Klein and G. G. Amatucci, J. Electrochem. Soc., 2006, 153, A159 CrossRef CAS.
- L. Zhang, D. Dambournet, A. Iadecola, D. Batuk, O. J. Borkiewicz, K. M. Wiaderek, E. Sakager, M. Shao, G. Chen and J.-M. Tarascon, Chem. Mater., 2018, 30, 5362 CrossRef CAS.
- Y.-Y. Hu, Z. Liu, K.-W. Nam, O. J. Borkiewicz, J. Chung, X. Hua, M. T. Dunstan, X. Yu, K. M. Wiaderek, L.-S. Du, K. W. Chapman, P. J. Chupas, X.-Q. Yang and C. P. Grey, Nat. Mater., 2013, 12, 1130 CrossRef CAS PubMed.
- H.-F. Wang, C. Tang, B.-Q. Li and Q. Zhang, Inorg. Chem. Front., 2018, 5, 521 RSC.
- Y. Takeda, R. Kanno, Y. Tsuji and O. Yamamoto, J. Power Sources, 1983, 9, 325 CrossRef CAS.
- J. Hu, H. Li and X. Huang, Electrochem. Solid-State Lett., 2005, 8, A66 CrossRef CAS.
- L.-Y. Jiang, S. Xin, X.-L. Wu, H. Li, Y.-G. Guo and L.-J. Wan, J. Mater. Chem., 2010, 20, 7565 RSC.
- R. Koksbang and P. Norby, Electrochim. Acta, 1991, 36, 127 CrossRef CAS.
- B. R. Maddox, C. S. Yoo, D. Kasinathan, W. E. Pickett and R. T. Scalettar, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 144111 CrossRef.
- W. Ren, B. Li, W. Liang, C. Jin and Z. Zhang, J. Alloys Compd., 2014, 596, 69 CrossRef CAS.
- B. Li, Y. Chen, H. Wang, W. Liang, G. Liu, W. Ren, C. Li, Z. Liu, G. Rao, C. Jin and Z. Zhang, Chem. Commun., 2014, 50, 799 RSC.
- S. Huang, X. Wu, J. Niu and S. Qin, RSC Adv., 2018, 8, 24561 RSC.
- J. Kim and A. Manthiram, J. Electrochem. Soc., 1997, 144, 3077 CrossRef CAS.
- K. Mukai and I. Yamada, Inorg. Chem. Front., 2018, 5, 1941 RSC.
- T. Uyama, K. Mukai and I. Yamada, RSC Adv., 2018, 8, 26325 RSC.
- K. Mukai, T. Uyama and I. Yamada, ACS Omega, 2019, 4, 6459 CrossRef CAS PubMed.
- T. Uyama, K. Mukai and I. Yamada, Inorg. Chem., 2019, 58, 6684 CrossRef CAS PubMed.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15 CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- T. Ohzuku and Y. Makimura, Chem. Lett., 2001, 7, 744 CrossRef.
- N. Yabuuchi and T. Ohzuku, J. Power Sources, 2003, 199–121, 171 CrossRef.
- S. Deng, Y. Zhang, D. Xie, L. Yang, G. Wang, X. Zheng, J. Zhu, X. Wang, Y. Yu, G. Pan, X. Xia and J. Tu, Nano Energy, 2019, 58, 355 CrossRef CAS.
- B. Liu, R. Fang, D. Xie, W. Zhang, H. Huang, Y. Xia, X. Wang, X. Xia and J. Tu, Energy Environ. Mater., 2018, 1, 196 CrossRef.
- S. Liu, X. Xia, S. Deng, D. Xie, Z. Yao, L. Zhang, S. Zhang, X. Wang and J. Tu, Adv. Mater., 2019, 31, 1806470 CrossRef PubMed.
- PDF-4+: Powder Diffraction File; International Centre for Diffraction Data: Newtown Square, PA, 2004.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of the pristine CrO2 sample, rietveld results of the pristine and HP/HT-treated CrO2 samples, V as a function of x in CrO2−xFx, SEM images of the x = 0.2 and 0.3 samples, EPMA analyses of the x = 0.20.2 samples, cycle performance at 318 K of the HP/HT-treated CrO2 sample, cycle performances at 318 K of the x = 0.05, 0.15, 0.2, and 0.3 samples, and XRD patterns of the x = 0.2 sample in the fully discharge state and after the cycle test at 318 K. See DOI: 10.1039/c9qi00765b |
| This journal is © the Partner Organisations 2019 |