
Interrogation of fractional crystallization behavior of a newly exploited chiral resolution method for racemic 1-(pyridin-2-yl)ethylamine via DFT-D3 calculations of cohesive energy†
Juhyun
Cho
 a,
Jong Hwa
Jeong
*a,
Myung Won
Lee
*b and
Youn K.
Kang
*c
a,
Jong Hwa
Jeong
*a,
Myung Won
Lee
*b and
Youn K.
Kang
*c
aDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: jeongjh@knu.ac.kr
bDepartment of Chemistry, Pukyong National University, Busan 48513, Republic of Korea. E-mail: mwlee@pknu.ac.kr
cDepartment of Chemistry, Sangmyung University, Seoul 03016, Republic of Korea. E-mail: younkang@smu.ac.kr
First published on 11th July 2019
Abstract
A novel chiral resolution method for racemic 1-(pyridin-2-yl)ethylamine (PEA) was developed in this study. Sequential conversion of 1-(pyridin-2-yl)ethylamine to 1-(pyridin-2-yl)-N-((4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine (PIC) and to dichloro{(E)-1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine}zinc (PIC-Zn) allowed for the spontaneous separation of dichloro{(1R,E)-1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine}zinc (RE-PIC-Zn) and dichloro{(1S,E)-1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine}zinc (SE-PIC-Zn) by fractional crystallization. The overall yields of both (S)- and (R)-PEA were 24.8% with estimated ee values of 98 and 99%, respectively. The preferred formation of diastereopure crystals over that of diastereomeric mixture crystals was interrogated by comparing the cohesive energies (Ecoh) of the respective crystals, as obtained via DFT calculations under periodic boundary conditions with a plane wave basis set based on the experimentally determined X-ray crystallographic structure. The Ecoh values of diastereopure SE-PIC-Zn and RE-PIC-Zn were −45.8 and −44.4 kcal mol−1, respectively. The Ecoh values of the diastereomeric mixture crystal (RE-PIC-Zn·SE-PIC-Zn·2CHCl3) and its virtual counterpart lacking CHCl3 (RE-PIC-Zn·SE-PIC-Zn) were −56.0 and −37.2 kcal mol−1, respectively. These data provided the background underlying the spontaneous segregation of the diastereopure crystals where the energy preference for forming diastereopure crystals over their corresponding diastereomeric mixture crystals is ca. 8 kcal mol−1.
1. Introduction
Typically, only one enantiomer of a chiral drug is physiologically active toward the biological target because biomacromolecules are constructed using L-amino acids as building blocks.1 This necessitates chiral resolution in the pharmaceutical industry first and across many fields of chemistry.2,3 Since the demonstration of the first chiral resolution by Louis Pasteur,4 the development of production processes to generate or isolate pure enantiomers has been remarkable.1–3,5–21 The numerous methods for producing pure enantiomers can be divided into two main approaches: (A) the chiral approach that aims for the selective synthesis of a single enantiomer and (B) the racemic approach that provides separation methods for chiral mixtures.1 The latter has attracted significant attention recently due to the progress in crystallization-based separation techniques.The propensity to form conglomerates of pure enantiomers rather than racemic crystals would be desirable for industrial scale enantioseparation, a process known as preferential crystallization.22–28 However, the probability of this type of behavior occurring is only 5–10%,5 hampering the wide application of this approach as a major enantioseparation method. The fractional crystallization of diastereomers that specifically implies spontaneous segregation into enantiopure diastereomers rather than forming diastereomeric mixtures is more complex compared to preferential crystallization of a conglomerate system. This may be a good alternative for outnumbering racemates provided that chemical conversion of those racemates into their corresponding diastereomers is easily accessible.29–31 Development of a novel protocol enabling fractional crystallization is thus indispensable.
Our group has extensively investigated asymmetric catalysis with a variety of metal complexes using newly synthesized chiral ligands.32–36 During the search for a chiral ligand, we encountered optically active 1-substituted-1-(pyridyl)methylamines. They are naturally occurring bioactive compounds that include tobacco alkaloids (i.e. nicotine, nornicotine, and anatabine)37–41 and histamine H1-receptor agonists.42–44 However, their potential as building blocks for the chiral ligand pool has attracted more of our attention. In this study, we focus on the chiral resolution of 1-(pyridin-2-yl)ethylamine, hereafter referred to as PEA, first reported in 1973 by Smith et al.45 by way of a classical approach using (+)-tartaric acid and only the (S)-form was obtained with an ∼9.5% overall yield. The enantiomeric excess (ee) of (S)-PEA was later confirmed to be 99% based on HPLC analysis of the (S)-PEA/(+)-tartaric acid salt.46 In 1977, Westley et al. separated (R)-PEA in a higher yield (37%) with much lower ee (78%) using lasalocid as a chiral derivatizing agent (CDA).47 In 1997, Gotor et al. separated both (R)- and (S)-PEA in 50% conversion with a moderately high ee (90 and 91% for (R)- and (S)-PEA, respectively) via enzyme catalyzed enantioselective aminolysis.48 This approach was later improved by McEachern et al., achieving a 52% yield with 75% ee for (R)-PEA and 41% yield with 99% ee for the corresponding (S)-enantiomer.49 On the other hand, enzyme catalyzed asymmetric synthesis from racemic 1-pyridylethylalcohol afforded enantiopure (R)- and (S)-PEA in >96% yield with ∼99% ee.50
The primary objective of this study was to develop a novel experimental protocol for the chiral resolution of PEAvia fractional crystallization. A classical approach was used for racemic PEA: treatment with CDA and subsequent separation. However, the interaction motif between the racemate and CDA was not a simple electrostatic interaction but formation of a strong N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C imino bond via condensation between the primary amine of PEA and the ketone group of (+)-camphor. Moreover, secondary metal coordination of the generated Schiff base ligand to a ZnCl2 moiety was devised to ensure solidification. The fractional crystallization conditions of spontaneous segregation into diastereopure (S,R,R)- and (R,R,R)-diastereomers were serendipitously obtained with ca. 60% overall yield and >98% diastereomeric excess (de). Details of the separation procedures are provided later in this manuscript.
C imino bond via condensation between the primary amine of PEA and the ketone group of (+)-camphor. Moreover, secondary metal coordination of the generated Schiff base ligand to a ZnCl2 moiety was devised to ensure solidification. The fractional crystallization conditions of spontaneous segregation into diastereopure (S,R,R)- and (R,R,R)-diastereomers were serendipitously obtained with ca. 60% overall yield and >98% diastereomeric excess (de). Details of the separation procedures are provided later in this manuscript.
Another objective was to elucidate the background of the fractional crystallization behavior of the generated diastereomeric systems quantitatively. We adopted the benchmark calculation protocol for non-covalent interactions in solids via DFT calculations under periodic boundary conditions (PBCs) with dispersion corrections (PBE-D3).51 This was made possible by the successful growth of three different molecular crystals, diastereopure (SRR)- and (RRR)-diastereomers and their mixture (SRR/RRR). We computed the cohesive energies of the respective crystal systems, representing the experimental sublimation energies.52 The calculated cohesive energies between the diastereopure and the diastereomeric mixture differed significantly (7.4–11.4 kcal mol−1), exceeding the chemical accuracy target (∼1 kcal mol−1) and explaining the fractional crystallization behavior, even considering the errors in the calculation. In addition to the cohesive energies, we computed the crystallization energies of both diastereopure (SRR)- and (RRR)-diastereomers, obtaining the zero-point energy exclusive electronic energy difference between solid and solution states to explain the preferred formation of the (S,R,R)-diastereomer over that of its (R,R,R)-counterpart. These results provide useful insights into the fractional crystallization behavior by providing quantitative measures as a benchmark for the future design of crystallization-based chiral resolution methods.
2. Experimental
2.1. Materials
All manipulations were carried out under air unless otherwise noted. Reagent grade toluene, dichloromethane, chloroform, methanol, and absolute ethanol were purchased from Aldrich and used without further purification. THF was purchased from Aldrich and was dried over Na/benzophenone ketyl and subsequently distilled from these reagents under nitrogen prior to use. Zinc chloride, sodium cyanide, and p-toluenesulfonic acid were purchased from Aldrich and were used without further purification.2.2. Instrumentation
Elemental analysis (C, H, and N) of the synthesized metal complexes was performed using an elemental analyzer (EA 1108; Carlo-Erba, Milan, Italy). The NMR spectra were recorded using a Bruker Avance III 500 NMR spectrometer. Chemical shifts of the 1H NMR spectra are relative to residual protons in the deuterated solvent (CDCl3, δ = 7.24). All coupling constants are reported in Hz. The chemical shifts of the 13C NMR spectra were measured relative to CDCl3 (77.00 ppm). Infrared (IR) spectra were recorded using a Bruker Alpha FT-IR spectrometer and the data are reported in reciprocal centimeters (cm−1). Circular dichroism (CD) spectra were recorded on a Jasco 715 spectropolarimeter.2.3. X-ray crystallographic analysis
Single crystals were mounted in a thin-walled glass capillary on an Enraf-Nonius CAD-4 diffractometer at 25 °C using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Unit cell parameters were determined by least-squares analysis of 25 reflections. The intensity data were collected in the ω/2θ scan mode and three standard reflections were monitored every 1 h during data collection. The data were corrected for Lorentz-polarization effects and decay and empirical absorption corrections with ψ-scans were applied. The structure was solved using the Patterson method and refined by full-matrix least squares techniques on F2 using the SHELXS and SHELXL programs of SHELX-97 package.53,54 The absolute structures were confirmed by anomalous dispersion effects with Friedel pairs which were not merged. All non-hydrogen atoms were refined anisotropically except for the disordered atoms. All hydrogen atoms were refined and positioned geometrically using the riding model with fixed isotropic thermal factors. The crystallographic data and the result of refinements are summarized in Table 1. The CCDC reference numbers are 1914650–1914652.†| SE-PIC-Zn | RE-PIC-Zn | 0.5RE-PIC-Zn·0.5SE-PIC-Zn·CHCl3 | |
|---|---|---|---|
| Empirical formula | C17H24Cl2N2Zn | C17H24Cl2N2Zn | C34H48Cl4Zn2N4·2CHCl3 |
| Formula weight | 392.65 | 392.65 | 1024.04 |
| Temperature (K) | 293(2) | 293(2) | 293(2) |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P212121 | P212121 | P21 |
| Unit cell dimensions | |||
| a (Å) | 9.7176(8) | 11.1816(9) | 12.7939(15) |
| b (Å) | 10.6577(6) | 12.7406(12) | 13.5872(10) |
| c (Å) | 18.0389(12) | 13.2577(13) | 13.4322(9) |
| α (°) | 90 | 90 | 90 |
| β (°) | 90 | 90 | 93.677(8) |
| γ (°) | 90 | 90 | 90 |
| Volume (Å3) | 1868.2(2), | 1888.7(3) | 2330.2(4) |
| Z | 4 | 4 | 2 |
| Density (calculated) (Mg m−3) | 1.396 | 1.381 | 1.46 |
| Absorption coefficient (mm−1) | 1.598 | 1.581 | 1.633 |
| F(000) | 816 | 816 | 1048 |
| Theta range for data collection (°) | 2.22 to 25.47 | 2.22 to 25.47 | 1.52 to 25.48 |
| Index ranges | −11 ≤ h ≤ −11 | −13 ≤ h ≤ −13 | −15 ≤ h ≤ 15 |
| −12 ≤ k ≤ −12 | −15 ≤ k ≤ −15 | −16 ≤ k ≤ 16 | |
| −21 ≤ l ≤ −21 | −16 ≤ l ≤ −16 | −16 ≤ l ≤ 16 | |
| Reflections collected | 4120 | 4053 | 9552 |
| Independent reflections | 3460 [R(int) = 0.0139] | 3496 [R(int) = 0.0174] | 8664 [R(int) = 0.0550] |
| Reflections observed (>2sigma) | 2739 | 3176 | 6739 |
| Data/restraints/parameters | 3460/0/203 | 3496/0/194 | 8664/1/474 |
| Goodness-of-fit on F2 | 1.036 | 1.175 | 1.139 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Final R indices [I > 2sigma(I)] | R 1 = 0.0262 | R 1 = 0.0347 | R 1 = 0.0497 |
| wR2 = 0.0648 | wR2 = 0.1163 | wR2 = 0.1640 | |
| R indices (all data) | R 1 = 0.0452 | R 1 = 0.0421 | R 1 = 0.0713 |
| wR2 = 0.0682 | wR2 = 0.1292 | wR2 = 0.1951 | |
| Absolute structure parameter | −0.016(14) | 0.04(2) | −0.018(19) |
| Largest diff. peak and hole (e Å−3) | 0.262 and −0.236 | 0.402 and −0.410 | 0.971 and −0.608 |
2.4. Syntheses
C), 164.37 (1C, pyridyl-C), 147.52–147.60 (1C, pyridyl-C), 135.46 (1C, pyridyl-C), 120.44–120.47 (1C, pyridyl-C), 119.68 (1C, pyridyl-C), 60.76–61.02 (1C, N–C), 52.63–52.70 (1C, camphor-C), 45.90 (1C, camphor-C), 42.89 (1C, camphor-C), 34.39–34.53 (1C, camphor-C), 31.11 (1C, camphor-C), 26.42–26.52 (1C, camphor-C), 21.90 (1C, N–C–CH3), 18.51 (1C, camphor-CH3), 18.01 (1C, camphor-CH3), 10.65–10.49 (1C, camphor-CH3). IR (liquid neat; cm−1): 3062(w), 2956(w), 2871(w), 1683(w), 1588(w), 1569(w), 1471(w), 1431(w), 1387(w), 1079(w), 748(w).
C), 158.48–158.58 (1C, pyridyl-C), 147.04–146.59 (1C, pyridyl-C), 139.79 (1C, pyridyl-C), 123.61 (1C, pyridyl-C), 121.48–121.60 (1C, pyridyl-C), 60.00–60.71 (1C, N–C), 55.63 (1C, camphor-C), 46.77–48.95 (1C, camphor-C), 42.07–42.55 (1C, camphor-C), 36.74–37.59 (1C, camphor-C), 29.82–31.12 (1C, camphor-C), 25.89–25.92 (1C, camphor-C), 23.08–23.73 (1C, N–C–CH3), 18.75–18.80 (1C, camphor-CH3), 17.85–18.02 (1C, camphor-CH3), 11.21–11.99 (1C, camphor-CH3). IR (solid neat; cm−1): 2963 (w), 1653 (w), 1608 (w), 1573 (w), 1483 (w), 1443 (w), 1373 (w), 1285 (w), 1261 (w), 1105 (w), 784 (w).
C), 159.44 (1C, pyridyl-C), 147.69 (1C, pyridyl-C), 140.73 (1C, pyridyl-C), 124.61 (1C, pyridyl-C), 122.49 (1C, pyridyl-C), 61.72 (1C, N–C), 56.65 (1C, camphor-C), 49.97 (1C, camphor-C), 43.08 (1C, camphor-C), 38.56 (1C, camphor-C), 30.81 (1C, camphor-C), 26.91 (1C, camphor-C), 24.07 (1C, N–C-CH3), 19.76 (1C, camphor-CH3), 19.03 (1C, camphor-CH3), 12.22 (1C, camphor-CH3). IR (solid neat; cm−1): 2967 (w), 1656 (w), 1608 (w), 1574 (w), 1485 (w), 1372 (w), 1287 (w), 1166 (w), 1053 (w), 783 (w). CD (CH3CN solution; nm): 235(−).
C), 159.61 (1C, pyridyl-C), 147.97 (1C, pyridyl-C), 140.86 (1C, pyridyl-C), 124.52 (1C, pyridyl-C), 122.54 (1C, pyridyl-C), 60.99 (1C, N–C), 57.35 (1C, camphor-C), 47.76 (1C, camphor-C), 43.55 (1C, camphor-C), 37.77 (1C, camphor-C), 32.13 (1C, camphor-C), 26.91 (1C, camphor-C), 24.73 (1C, N–C–CH3), 19.81 (1C, camphor-CH3), 18.85 (1C, camphor-CH3), 12.99 (1C, camphor-CH3). IR (solid neat; cm−1): 2958 (w), 1659(w), 1608 (w), 1574(w), 1485(w), 1444(w), 1374(w), 1288(w), 1160(w), 1104(w), 784(w). CD (CH3CN solution; nm): 236(+).
C), 165.37 (1C, pyridyl-C), 148.52 (1C, pyridyl-C), 136.47 (1C, pyridyl-C), 121.44 (1C, pyridyl-C), 120.69 (1C, pyridyl-C), 61.77 (1C, N–C), 53.71 (1C, camphor-C), 46.90 (1C, camphor-C), 43.91 (1C, camphor-C), 35.54 (1C, camphor-C), 32.12 (1C, camphor-C), 27.53 (1C, camphor-C), 23.01 (1C, N–C–CH3), 19.53 (1C, camphor-CH3), 19.04 (1C, camphor-CH3), 11.49 (1C, camphor-CH3). IR (liquid neat; cm−1): 3051 (w), 2955 (w), 2872 (w), 1683 (w), 1588 (w), 1470 (w), 1431 (w), 1388(w), 1047 (w), 764 (w), 748(w). CD (CH3CN solution; nm): 221(−), 238(+), 277(+).
C), 164.34 (1C, pyridyl-C), 147.59 (1C, pyridyl-C), 135.45 (1C, pyridyl-C), 120.46 (1C, pyridyl-C), 119.82 (1C, pyridyl-C), 61.01 (1C, N–C), 52.62 (1C, camphor-C), 45.97 (1C, camphor-C), 42.89 (1C, camphor-C), 34.37 (1C, camphor-C), 31.17 (1C, camphor-C), 26.42 (1C, camphor-C), 21.89 (1C, N–C–CH3), 18.51 (1C, camphor-CH3), 18.01 (1C, camphor-CH3), 10.65 (1C, camphor-CH3). IR (liquid neat; cm−1): 3064 (w), 2957(w), 2871(w), 1682 (w), 1588(w), 1568(w), 1471(w), 1431(w), 1387(w), 1120(w), 748(w). CD (CH3CN solution; nm): 223(+), 240(−), 275(−).
2.5. Computational details
The DFT calculations of the isolated molecules were performed using the Gaussian 16 program package (A.03 version).55 Geometry optimizations of the organic molecules (PIC) were performed using the PBE1PBE hybrid functional,56–58 while those of the metal complexes (PIC-Zn) were performed using the GGA functional with dispersion corrections (PBE-D3)58,59 for consistency with the PBC calculations which used the plane wave basis set. All calculations were performed with the Karlsruhe basis sets developed by Ahlrichs et al. (def2TZVP).60,61 For Zn, relativistic ECPs for the inner 10 electrons were represented by Stuttgart RSC 1997 (SDD).62–64 Geometry optimizations of all model compounds were performed both in the gas phase and in solvents. The solvent was modeled as a dielectric continuum using a self-consistent reaction field (SCRF)65–67 approach by a conductor-like polarizable continuum model (CPCM)68,69 in toluene (PIC) and methanol (PIC-Zn) as implemented in Gaussian 16.Vibrational frequency calculations were performed under the same solvent environment with the optimized geometries. The obtained information was used to calculate the free energy of the given molecule. Solvation energies were calculated using the thermodynamic parameters obtained from the optimized structures computed in the gas phase and dielectric continuum. Rotational barriers through NC double bonds in the PIC molecules that connect the 1,7,7-trimethylbicyclo[2.2.1]heptyl and 1-(pyridin-2-yl)ethylamine groups were estimated from the potential surface scan by varying the dihedral angle.
DFT calculations of the molecular crystals were performed using the VASP program (v5.4)70 with the PAW potentials.71,72 A plane wave basis set with a cutoff of 600 eV was used for the calculations. All calculations employed a 2 × 2 × 2 k-point mesh after confirming that finer k-point grids gave virtually the same result. The break condition for the electronic SCF loop was set to 10−4 eV, while that for the ionic relaxation was set to 10−3 eV. The PBE exchange–correlation functional with dispersion corrections (PBE-D3)58,59 was used, as implemented in VASP.73 The optimization process began from the atomic coordinates and lattice parameters of the experimentally determined X-ray structures. Both the atomic coordinates and lattice parameters were optimized to reach energy minima. The energy of the gas-phase molecule (Egas) was approximated by putting a single molecule in an enlarged unit cell (25 × 20 × 20 Å) to eliminate interactions between neighboring molecules for comparison with the results obtained from Gaussian 16. Based on the work by Moellmann and Grimme, the cohesive energy was calculated using the following relation:51
| Ecoh = Esolid/N − Egas |
| Ecryst = Esolid/N − Esol |
This relation specifically describes the dissolution energy with the opposite sign calculated using the zero-point exclusive electronic energy. It should be noted that Esolid was obtained by PBC calculations with a plane wave basis set implemented in the VASP program, while Esol was obtained via CPCM calculations with a Gaussian basis set (def2TZVP) implemented in the Gaussian 16 program. For the Gaussian calculations, the same PBE-D3 was used.
For calculations of the SE-PIC-Zn·RE-PIC-Zn·CHCl3 mixed crystal, Ecoh was computed by
| Ecoh = [Esolid − {2Egas (SE-PIC-Zn) + 2Egas (RE-PIC-Zn) + 4Egas (CHCl3)}]/4 |
The optimization of the virtual crystal SE-PIC-Zn·RE-PIC-Zn began from the experimentally determined atomic coordinates and lattice parameters after removing the 4 CHCl3 molecules. Ecoh was computed by
| Ecoh = [Esolid − {2Egas (SE-PIC-Zn) + 2Egas (RE-PIC-Zn)}]/4. |
3. Results and discussion
Inspired by the work of Pedro et al.,74,75 racemic PEA was converted into separable diastereomers, 1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine (PIC), by condensation with (1R)-(+)-camphor. In contrast to Pedro et al., p-toluenesulfonic acid was used as a catalyst, affording a high yield (94.7%). While four different diastereomers are anticipated to be formed, as illustrated in Scheme 1, the 1H NMR results indicated that only two isomers were present in the product mixture (Fig. 1). Two sets of multiplets, each indicative of the methine proton in the camphor group, distinctively resided at δ 2.33 and δ 2.47 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 intensity ratio. The multiplet in the δ 4.55–4.61 region was composed of two overlapping quartets, each characteristic of the methine proton at the carbon near the imino group. This product mixture was hardly separable via column chromatography, which required further investigation.
:1 intensity ratio. The multiplet in the δ 4.55–4.61 region was composed of two overlapping quartets, each characteristic of the methine proton at the carbon near the imino group. This product mixture was hardly separable via column chromatography, which required further investigation.
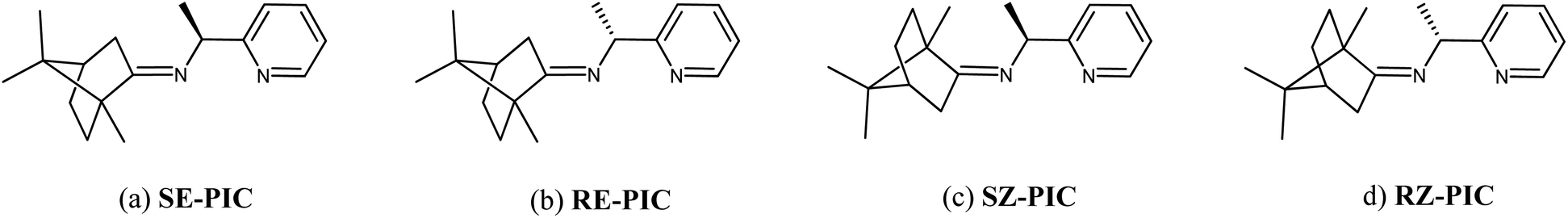 | ||
| Scheme 1 Possible structures of the 4 diastereomeric isomers of PIC. (a) (1S,E)-1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine (SE-PIC), (b) (1R,E)-1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine (RE-PIC), (c) (1S,Z)-1-(pyridin-2-yl)-N-((4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine (SZ-PIC), and (d) (1R,Z)-1-(pyridin-2-yl)-N-((4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine (RZ-PIC). | ||
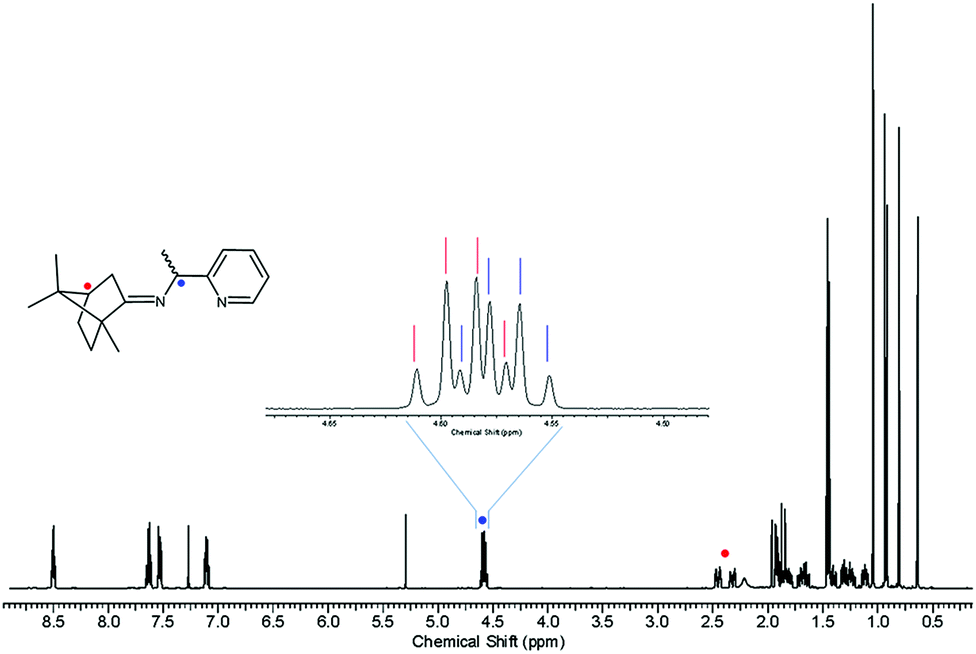 | ||
| Fig. 1 1H NMR spectrum of PIC. | ||
Recently, we reported the facile separation of the diastereo-chiral N,N′-diamine ligand via fractional crystallization and demetallation of the corresponding Zn(II) complex.76 Metallation of the diastereomeric mixture, containing (2R,3R)-N2,N3-bis((R)-1-phenylethyl)butane-2,3-diamine (RR) and (2S,3S)-N2,N3-bis((R)-1-phenylethyl)butane-2,3-diamine (SS), with ZnCl2 yielded the corresponding Zn(II) complexes, among which only RR was selectively separated by fractional crystallization. Considering this precedent, we attached the ZnCl2 moiety to the PIC mixture anticipating that the propensity for fractional crystallization could be generated by metal complexation. The reaction proceeded very smoothly and stirring the mixture of PIC and an equimolar amount of ZnCl2 in ethanol at ambient temperature afforded the desired product (PIC-Zn) in 94.4% yield. The Zn metallated diastereomeric mixture complexes were segregated by fractional crystallization under optimized conditions. MeOH was most effective as a crystallizing solvent. The detailed separation procedure described in the Experimental section is summarized in Scheme 2 and graphically illustrated in Fig. 2.
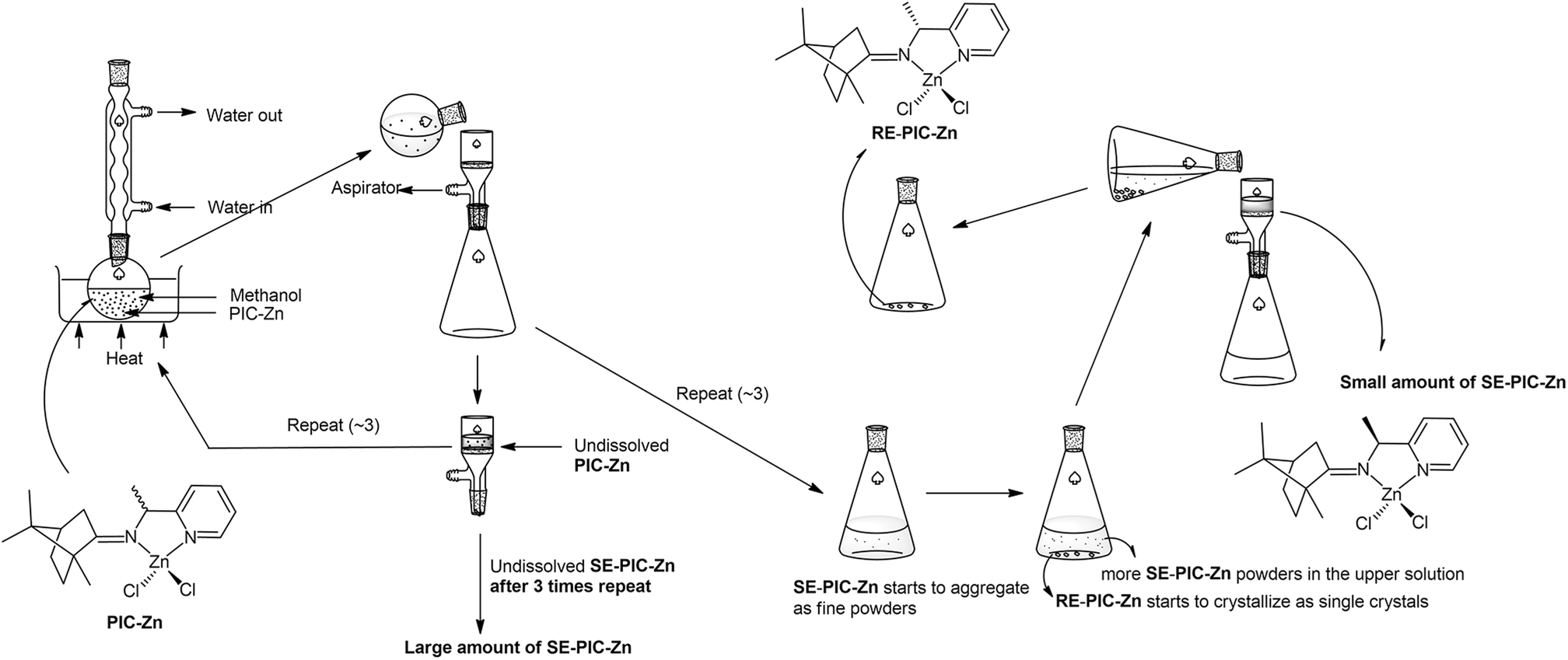 | ||
| Fig. 2 Schematic illustration of separation flow of the diastereomeric mixture PIC-Znvia fractional crystallization. See the Experimental section for details. | ||
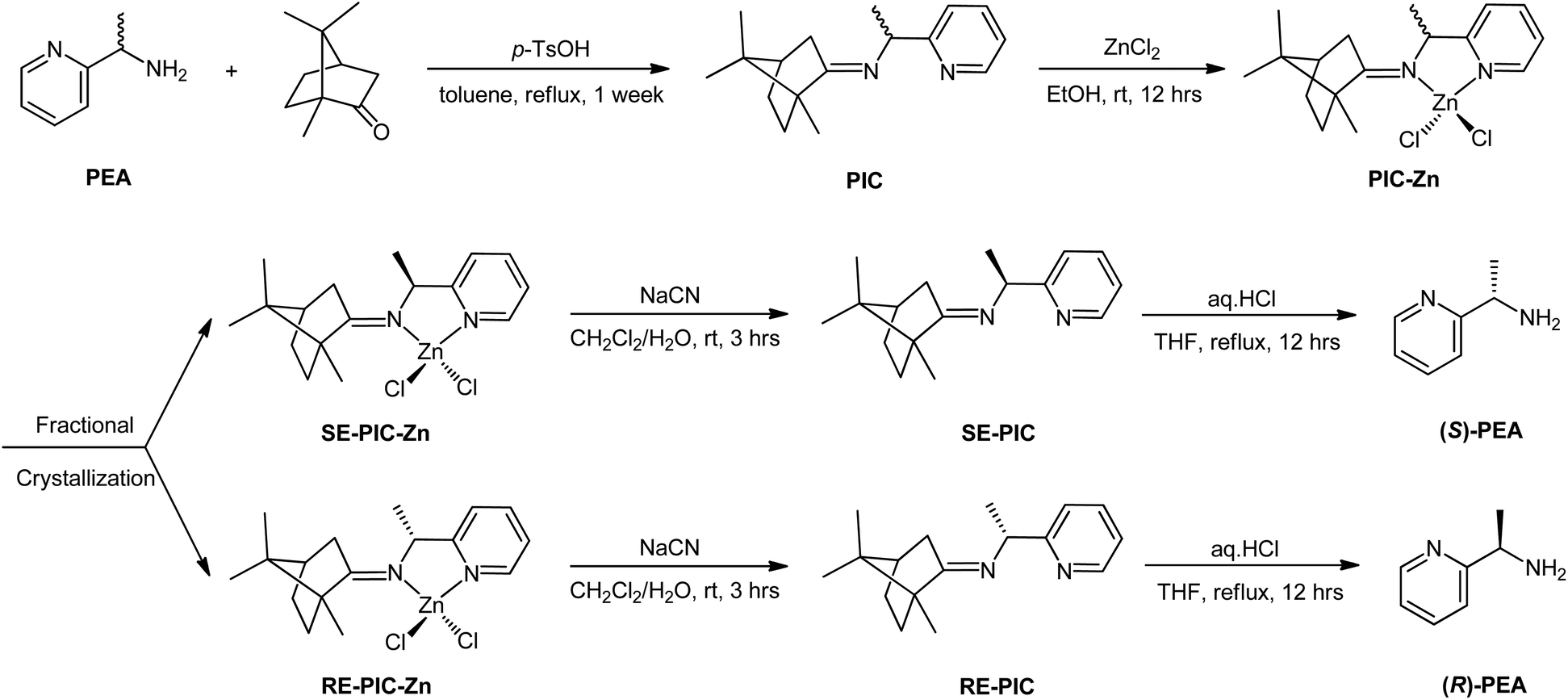 | ||
| Scheme 2 Separation scheme of PEA. | ||
Single crystals of RE-PIC-Zn suitable for X-ray crystallographic analysis were obtained via slow evaporation of a hot MeOH solution containing the compounds of interest to 25 °C. However, SE-PIC-Zn could not be grown in the same solvent system due to its low solubility and was separately grown via layering of n-hexane in CH2Cl2 solution. Crystal data, details regarding the data collection, and refinement parameters are listed in Table 1. The ORTEP drawings of the complexes are shown in Fig. 3 with atomic labelling for nonhydrogen atoms. Selected bond distances and angles are listed in Table 2. The crystal structure of the white precipitates formed during the cooling process of the hot filtrate was revealed as dichloro{(1S,E)-1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine}zinc (SE-PIC-Zn). The other compound was dichloro{(1R,E)-1-(pyridin-2-yl)-N-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethylamine}zinc (RE-PIC-Zn). Because the added (+)-camphor moiety was in the (R,R) configuration, the overall configurations of SE-PIC-Zn and RE-PIC-Zn were (S,R,R) and (R,R,R), respectively. It should be noted that both structures have the (E)-configuration for the imino NC double bond. As noted earlier, only two diastereomers instead of the possible four were confirmed by X-ray crystallography.
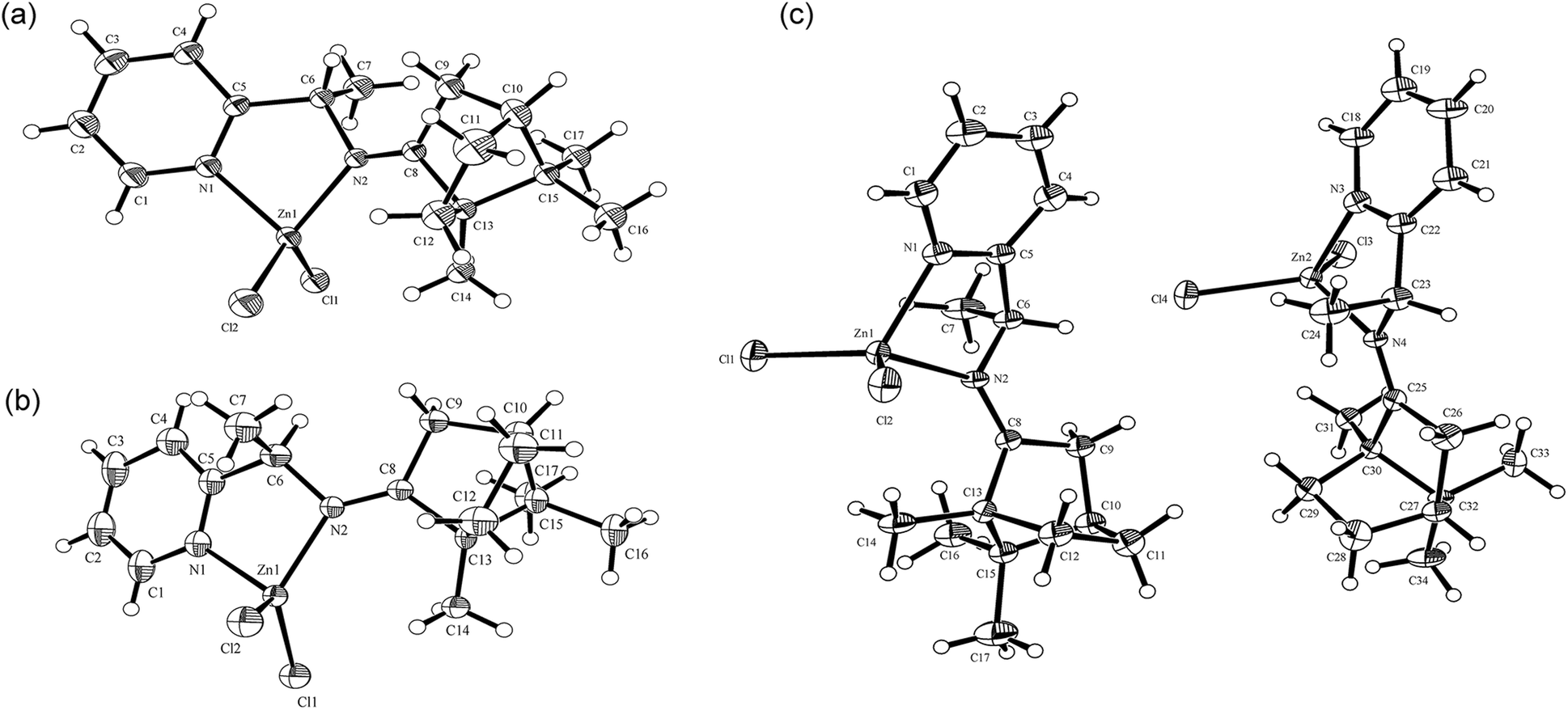 | ||
| Fig. 3 ORTEP drawings of (a) SE-PIC-Zn and (b) RE-PIC-Zn and (c) SE-PIC-Zn and RE-PIC-Zn from 0.5RE-PIC-Zn·0.5SE-PIC-Zn·CHCl3. CHCl3 molecules are not shown. | ||
| Crystal system | SE-PIC-Zn | RE-PIC-Zn | 0.5RE-PIC-Zn·0.5SE-PIC-Zn·CHCl3 | |
|---|---|---|---|---|
| Configuration | (S,R,R) | (R,R,R) | (S,R,R) | (R,R,R)a |
| a Numbering scheme for the RRR form in the 0.5RE-PIC-Zn·0.5SE-PIC-Zn·CHCl3 crystal system in Table 2 follows that of the RRS form with the corresponding bond lengths and angles instead of using that of the ORTEP diagram in Fig. 3. | ||||
| Zn(1)–N(1) | 2.056 | 2.052 | 2.059 | 2.060 |
| Zn(1)–N(2) | 2.063 | 2.076 | 2.056 | 2.090 |
| Zn(1)–Cl(1) | 2.217 | 2.207 | 2.207 | 2.210 |
| Zn(1)–Cl(2) | 2.208 | 2.216 | 2.211 | 2.210 |
| N(1)–C(5) | 1.344 | 1.325 | 1.346 | 1.320 |
| N(1)–C(1) | 1.338 | 1.341 | 1.335 | 1.344 |
| N(2)–C(8) | 1.265 | 1.264 | 1.303 | 1.260 |
| N(2)–C(6) | 1.485 | 1.478 | 1.449 | 1.482 |
| C(5)–C(6) | 1.500 | 1.511 | 1.533 | 1.512 |
| C(6)–C(7) | 1.549 | 1.545 | 1.533 | 1.536 |
| N(1)–Zn(1)–N(2) | 82.2 | 81.9 | 81.9 | 82.5 |
| N(1)–Zn(1)–Cl(1) | 107.8 | 102.7 | 113.1 | 105.3 |
| N(2)–Zn(1)–Cl(1) | 115.2 | 126.8 | 113.8 | 125.1 |
| N(1)–Zn(1)–Cl(2) | 108.6 | 115.8 | 105.5 | 113.7 |
| N(2)–Zn(1)–Cl(2) | 117.3 | 107.7 | 118.5 | 108.8 |
| Cl(1)–Zn(1)–Cl(2) | 118.6 | 116.6 | 117.8 | 116.4 |
| C(5)–N(1)–Zn(1) | 113.6 | 113.9 | 113.3 | 114.0 |
| C(1)–N(1)–Zn(1) | 126.8 | 126.4 | 126.9 | 127.2 |
| C(8)–N(2)–C(6) | 117.9 | 118.1 | 116.3 | 119.1 |
| C(8)–N(2)–Zn(1) | 130.8 | 133.1 | 132.8 | 133.5 |
| C(6)–N(2)–Zn(1) | 110.3 | 108.2 | 110.9 | 106.9 |
| N(1)–C(5)–C(6) | 118.0 | 117.0 | 116.7 | 116.2 |
| N(2)–C(6)–C(5) | 111.0 | 110.5 | 109.9 | 112.0 |
| N(2)–C(6)–C(7) | 107.8 | 109.3 | 110.0 | 107.9 |
| C(5)–C(6)–C(7) | 109.8 | 108.8 | 109.1 | 110.2 |
The dominant formation of the (E)-conformation in the condensation step deserves additional consideration. We performed DFT calculations for the four diastereomeric geometries of the PIC molecule. The relative free energies of the four diastereomers in the gas phase at 298.15 K were 0.14, 0.00, 6.22, and 6.42 kcal mol−1 for SE-, RE-, SZ-, and RZ-PIC, respectively. The values calculated in the dielectric continuum of toluene were −0.03, 0.00, 6.62, and 6.45, respectively, which are similar to those obtained in the gas phase. Both results clearly indicate that the PIC molecule prefers the E-conformation for the imino NC double bond by ca. 6 kcal mol−1. This energy difference is significant and corresponds to a ca. 25000-fold excess of the E-conformation over the Z-counterpart. The estimated rotational barrier between the E- and Z-conformations is ∼24 kcal mol−1, indicating that the condensation reaction for two weeks at the boiling temperature of toluene should be sufficient to shift the equilibrium toward the E-conformation whether the chiral center is S or R.
The overall geometries of both SE-PIC-Zn and RE-PIC-Zn were determined to be an ordinary distorted tetrahedron composed of a ZnL2Cl2-type skeleton with larger Cl(1)–Zn–Cl(2) angles (118.6° and 116.6° for SE-PIC-Zn and RE-PIC-Zn, respectively) and smaller N(1)–Zn–N(2) angles (82.2° and 81.9°, respectively). The lengths of the corresponding bonds between the two structures were very similar with a maximum difference of 0.024 Å for the C1–C2 bond. However, notable differences in the bond angles were found for the angles containing a Zn metal center. For example, the N(2)–Zn–Cl(1) angle of SE-PIC-Zn was 115.2° while that of RE-PIC-Zn was 126.8°, a difference of 11.6°.
The crystal system and space group of the two structures are both orthorhombic and non-centrosymmetric P212121, respectively, as the most abundant form for the enantiopure chiral molecular crystals. The unit cell dimensions, a, b, and c, differed dramatically: 9.7176, 10.6577, and 18.0389 Å for SE-PIC-Zn and 11.1816, 12.7406, and 13.2577 Å for RE-PIC-Zn, respectively (Fig. 4). The overall shape of the unit cell of RE-PIC-Zn is close to cubic while that of SE-PIC-Zn is elongated tetragonal. Both unit cells contain four molecules of the corresponding diastereomers. The overall cell volumes were similar to each other: 1868.2 and 1888.7 Å3 for SE-PIC-Zn and RE-PIC-Zn, respectively, leading to similar densities (1.396 and 1.381 g cm−3).
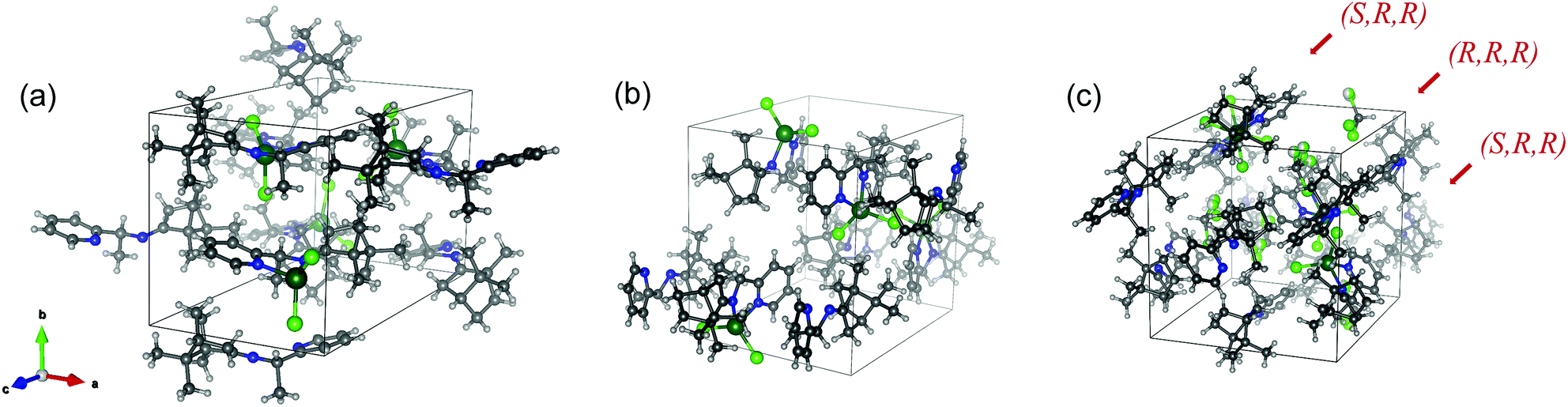 | ||
| Fig. 4 Unit cells of (a) SE-PIC-Zn, (b) RE-PIC-Zn, and (c) 0.5RE-PIC-Zn·0.5Se-PIC-Zn·CHCl3. | ||
The diastereomeric excess of the separated diastereomers was determined by 1H NMR. The methine region (C6) of the 1H NMR spectra is shown in Fig. 5, showing 98 and 99% of diastereomeric excess for SE-PIC-Zn and RE-PIC-Zn, respectively, indicating that fractional crystallization segregated two diastereomers almost completely.
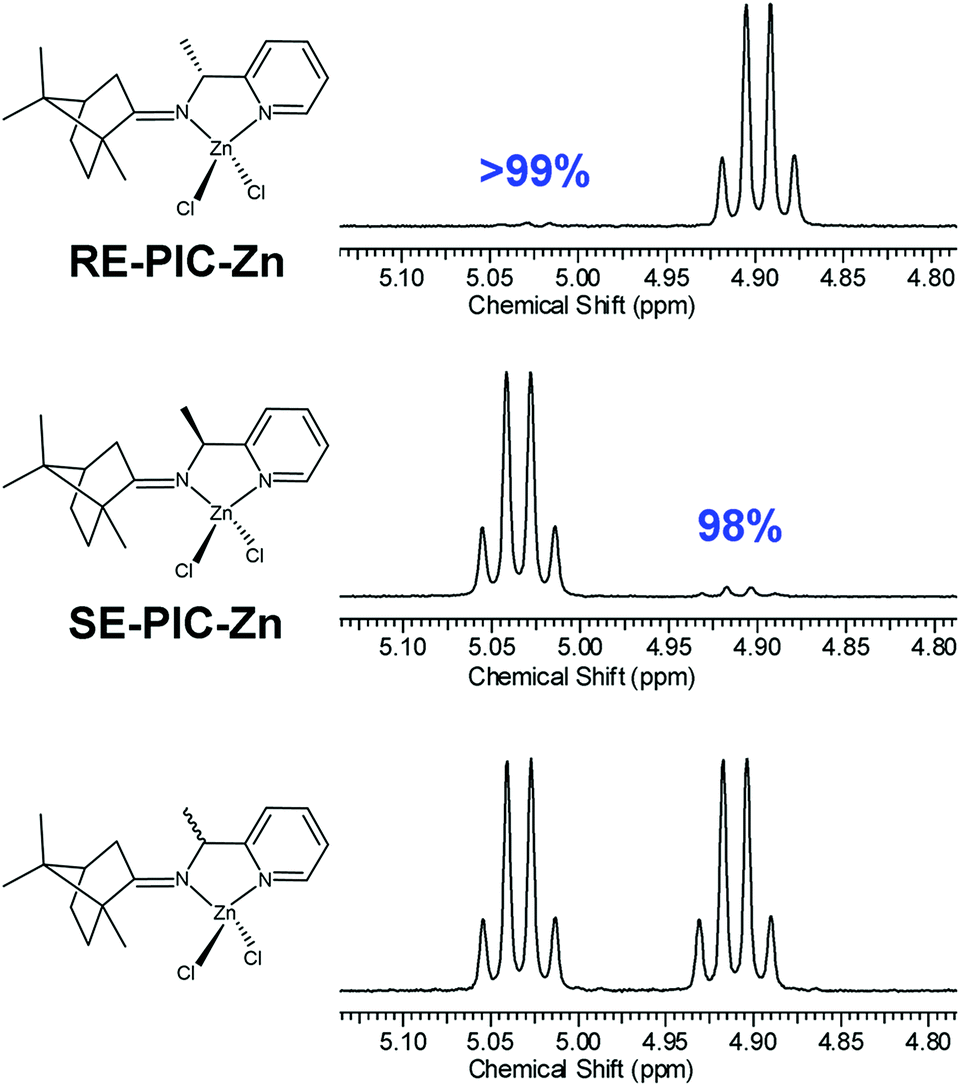 | ||
| Fig. 5 Methine region 1H NMR spectra of diastereopure RE-PIC-Zn (top), SE-PIC-Zn (middle) and the diastereomeric mixture PIC-Zn (bottom). The detailed conformation of NC double bonds of both complexes was determined by X-ray crystallography. | ||
The CDA removal processes were straightforward and smooth. The ZnCl2 moiety in the RE-PIC-Zn (or SE-PIC-Zn) was easily removed by treatment with aqueous NaCN in dichloromethane solution at ambient temperature, affording a virtually quantitative yield (96.7%). The separated RE-PIC and SE-PIC were analyzed by 1H NMR, 13C NMR, and IR spectroscopic methods. The 1H NMR spectra of the demetallated species are shown in Fig. 6. The camphor group in the RE-PIC (or SE-PIC) was removed by hydrolysis: THF solution of RE-PIC (or SE-PIC) in the presence of aqueous HCl was refluxed for 12 h, affording an ∼93% yield of the desired product. The overall yields of the obtained pure (S)- and (R)-PEA were both 24.2%. The circular dichroism (CD) spectra of the separated diastereomers, RE-PIC/SE-PIC and RE-PIC-Zn/SE-PIC-Zn, as well as enantiomers, R-PEA/S-PEA, were measured in acetonitrile (Fig. 7). The CD spectra of the enantiomeric couple were found to be mirror images of each other. R-PEA exhibited positive and negative Cotton effects at 243 and 269 nm, respectively. For RE-PIC/SE-PIC diastereomers, CD spectra were not mirror images of each other, although the regions of the Cotton effect were similar to each other. RE-PIC exhibited a positive Cotton effect at 238 nm and negative ones at 221 and 277 nm. SE-PIC showed a positive Cotton effect at 223 and 275 nm and a negative one at 240 nm. Interestingly, only the intensity of the negative Cotton effect observed in SE-PIC was a factor of two smaller than that of the positive Cotton effect of RE-PIC. In the case of RE-PIC-Zn/SE-PIC-Zn diastereomers, CD spectra were fortuitously mirror images of each other. The positive/negative Cotton effects of RE-PIC-Zn/SE-PIC-Zn were observed at 234/235 nm, respectively. The intensity of the positive Cotton effect in RE-PIC-Zn was a bit smaller than that of the negative Cotton effect in SE-PIC-Zn.
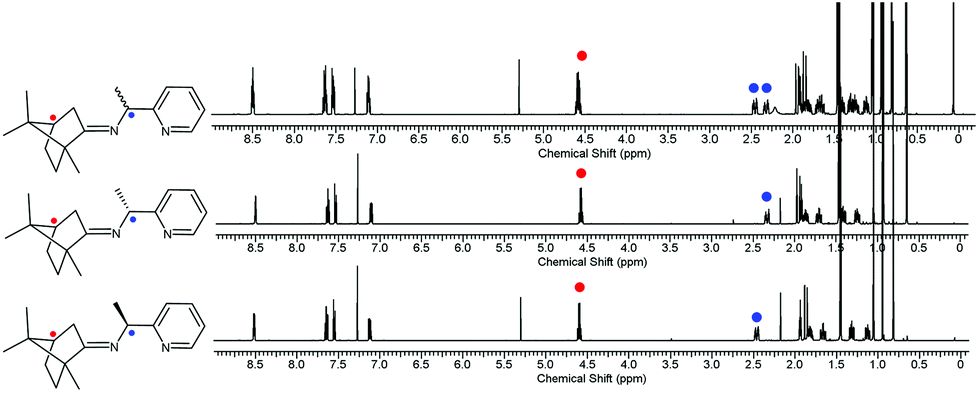 | ||
| Fig. 6 1H NMR spectra of the diastereomeric mixture of PIC (top), diastereopure RE-PIC (middle), and SE-PIC (bottom). | ||
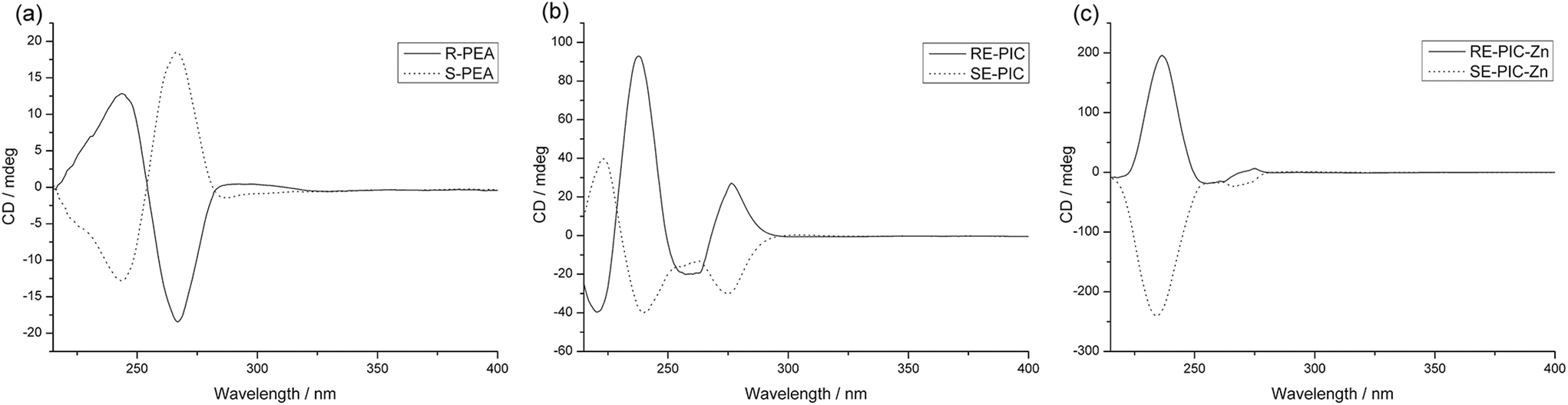 | ||
| Fig. 7 Observed CD spectra of (a) R-PEA (solid) and S-PEA (dotted) (c = 1.0 mM), (b) RE-PIC (solid) and SE-PIC (dotted) (c = 2.0 mM), and (c) RE-PIC-Zn (solid) and SE-PIC-Zn (dotted) (c = 2.0 mM) in CH3CN. | ||
During the fractional crystallization tests, we obtained single crystals that contained both SE-PIC-Zn and RE-PIC-Zn in the same unit cell by layering n-hexane over the CHCl3 solution of the SE-PIC-Zn and RE-PIC-Zn mixture. The crystal data, details of the data collection, and refinement parameters for the diastereomeric mixture crystal are listed in Table 1, and the ORTEP drawings of the complexes are shown in Fig. 3 with the atomic labeling for nonhydrogen atoms. Selected bond distances and angles are listed in Table 2. The structural parameters and characteristic features found for the SE-PIC-Zn·RE-PIC-Zn·2CHCl3 crystal are similar to those of the corresponding diastereopure crystals. Interestingly, if not very unusually, equal numbers of additional solvent molecules were observed in the unit cell. The total number of molecules in the unit cells was 8: 2 SE-PIC-Zn, 2 RE-PIC-Zn, and 4 CHCl3. The unit cell dimensions increased due to the crowding and were 12.79, 13.59, 13.43 Å for a, b, and c, respectively, forming a nearly cubical cell shape. The cell volume was 2330.2 Å, which is ca. 25% larger than that of the diastereopure crystal.
The experimentally determined 3-dimensional structures of both diastereopure and diastereomeric mixture crystals allowed for the subsequent DFT calculations to elucidate the underlying background of the observed fractional crystallization behavior. Specifically, we attempted to provide an explanation for the generation of diastereopure crystals in a MeOH solution of a diastereomeric mixture and a diastereomeric mixture crystal in the corresponding CHCl3 solution. The calculation was performed to obtain accurate energies of the molecular crystals and further details regarding the calculation criteria are now discussed. For calculations of molecular crystals, it is important to describe the long-range London dispersion interactions. Recent progress in DFT has overcome its intrinsic lack of dispersion interactions (e.g., DFT-D3 by Grimme59,77–79 and DFT + vdW by Tkatchenko80–82) which include the pairwise interaction. An improvement by considering the many body dispersions (MBDs) that mainly correct for the overestimation of lattice energies by pairwise interactions was later made.83–86 According to Grimme et al. who tested the calculation protocols with a 23 benchmark test set, the mean absolute error of the MBD approach was larger than that of the pairwise approach when the PBE functional was used.51 Therefore, we did not consider the MBD effect. For selecting an exchange–correlation functional, it is largely accepted that a hybrid functional exhibits better performance compared to a GGA. Grimme's report indicates that the performance difference between the hybrid PBE0 and GGA PBE functionals is a maximum of only 0.5 kcal mol−1.51 Our test results for molecular crystals showed that a longer computational time of nearly an order of magnitude was required when the PBE0 functional was used compared to that of PBE. Thus, DFT-D3 with the PBE exchange–correlation functional was used to obtain the cohesive energies (Ecoh) of the molecular crystals to evaluate their relative lattice energies. For consistency, we also used the PBE functional for the Gaussian calculations.
The calculated lattice parameters of the four crystal systems are summarized in Table 3. The calculated crystal structures of SE-PIC-Zn, RE-PIC-Zn, and RE-PIC-Zn·SE-PIC-Zn·2CHCl3 were well reproduced by optimization. The calculated cell dimensions were slightly smaller than experimentally determined dimensions, indicating that the dispersion interaction was slightly overestimated.
| SE-PIC-Zn | RE-PIC-Zn | 0.5RE-PIC-Zn·0.5SE-PIC-Zn·CHCl3 | 0.5RE-PIC-Zn·0.5SE-PIC-Zn | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Calc. | Error (%) | Exp. | Calc. | Error (%) | Exp. | Calc. | Error (%) | Exp. | Calc. | |
| a (Å) | 9.65 | 0.7 | 9.72 | 10.97 | 1.9 | 11.18 | 12.56 | 1.9 | 12.79 | 12.33 |
| b (Å) | 10.39 | 2.5 | 10.66 | 12.50 | 1.9 | 12.74 | 13.44 | 1.1 | 13.59 | 13.23 |
| c (Å) | 17.80 | 1.3 | 18.04 | 13.17 | 0.7 | 13.26 | 13.45 | 0.1 | 13.43 | 13.29 |
| α (°) | 90.00 | 0 | 90.00 | 90.00 | 0 | 90.00 | 90.00 | 0 | 90.00 | 90.00 |
| β (°) | 90.00 | 0 | 90.00 | 90.00 | 0 | 90.00 | 93.83 | 0.2 | 93.68 | 94.82 |
| γ (°) | 90.00 | 0 | 90.00 | 90.00 | 0 | 90.00 | 90.00 | .0 | 90.00 | 90.00 |
| V (Å3) | 1786.0 | 4.4 | 1868.2 | 1805.4 | 4.4 | 1888.7 | 2262.3 | 2.9 | 2330.2 | 2159.4 |
The calculated Ecoh values were −45.8 and −44.4 kcal mol−1 for SE-PIC-Zn and RE-PIC-Zn, respectively (Fig. 8), representing largely similar values with that of SE-PIC-Zn being only slightly lower by 1.4 kcal mol−1. However, the Ecoh of RE-PIC-Zn·SE-PIC-Zn·2CHCl3 was −56.0 kcal mol−1, which was approximately 10 kcal mol−1 more negative compared to that of the diastereopure crystals. This cohesive energy is actually for the 0.5SE-PIC-Zn/0.5RE-PIC-Zn/CHCl3 molecular composition, which is somewhat different from those of the pure SE-PIC-Zn and RE-PIC-Zn in which contribution from CHCl3 is absent. The additional CHCl3 molecules likely acts as an adhesive, increasing the overall lattice energy. This result explains the favored formation of the diastereomeric mixture in the presence of CHCl3.
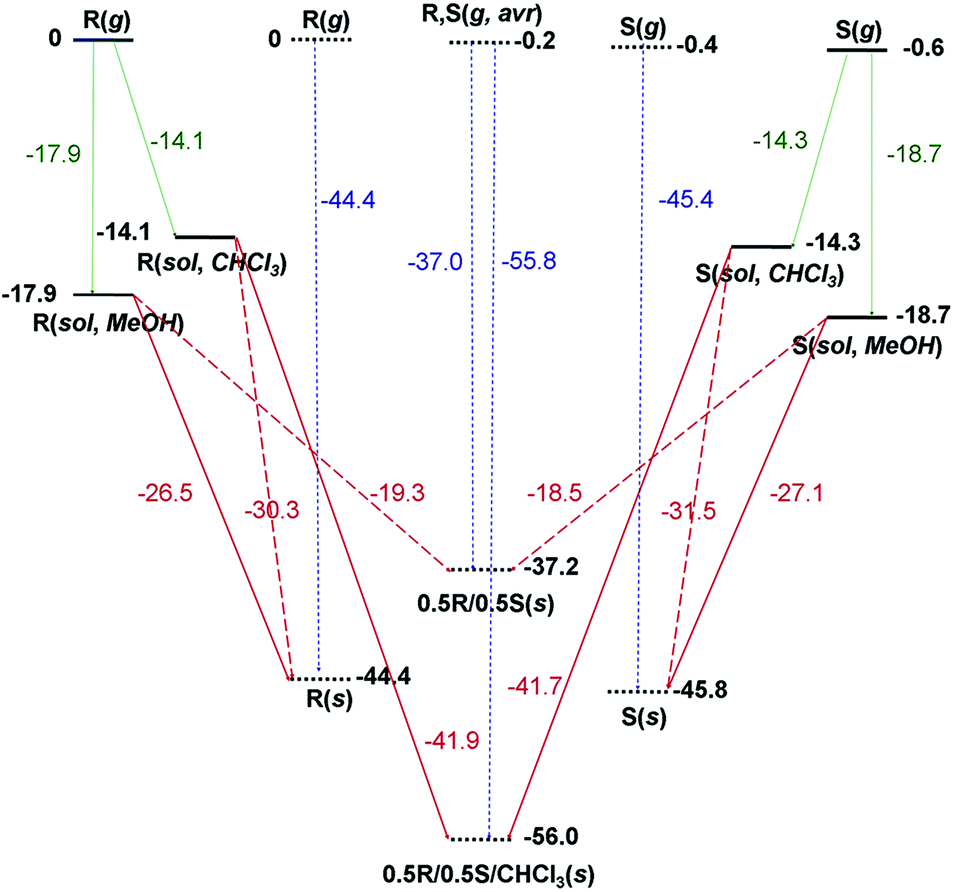 | ||
| Fig. 8 Calculated energy profile of the gas phase (g), solution phase (sol) with a designated solvent, and solid phase (s) of RE-PIC (R) and SE-PIC (S). Stationary states are represented as thick solid lines (isolated molecules) or thick dotted lines (molecular crystals). Numbers in bold type beside the state lines are zero-point exclusive electronic energies of states. Numbers in normal type beside the arrows are energy differences between two states, which represent solvation energies (green), crystallization energies (the opposites of dissolution energies, dark red), and cohesive energies (blue). | ||
On the other hand, the Ecoh value for the crystal composed only of two diastereomers was computed from the crystal structure obtained by optimization after removing four CHCl3 molecules manually. The cell angles of the optimized SE-PIC-Zn·RE-PIC-Zn crystal were well preserved and the monoclinic system was maintained. The lattice parameters were slightly reduced after the removal of the CHCl3 molecules and the cell volume was reduced by 4.6%. The Ecoh value was −37.2 kcal mol−1, which is 7.2 and 8.6 kcal mol−1 more positive than those of SE-PIC-Zn and RE-PIC-Zn, respectively. This is very interesting in two respects; although the energy of the SE-PIC-Zn·RE-PIC-Zn crystal system was obtained from the local minimum around the RE-PIC-Zn·SE-PIC-Zn·2CHCl3 structure, which is most likely a metastable diastereomeric mixture state, the degree of destabilization of Ecoh relative to those of pure SE-PIC-Zn and RE-PIC-Zn was similar to that of the stabilization of Ecoh originating from RE-PIC-Zn·SE-PIC-Zn·2CHCl3. In addition, the amount of destabilization energy was well beyond the coveted “chemical accuracy” target (1 kcal mol−1) and likely much larger than the error range derived from the misleading calculation protocol, if any, based on the previously published reports.51 Therefore, it is not unreasonable to speculate that the formation of diastereopure crystals, either SE-PIC-Zn or RE-PIC-Zn, is favorable compared to that of the diastereomeric mixture crystal by ca. 8 kcal mol−1.
We also considered the fractional crystallization behavior in terms of crystallization energy point of view. In this manuscript, the crystallization energy specifically implies the electronic energy difference between the solid state and the solution state. As shown in Fig. 8, the MeOH solution of both SE-PIC-Zn and RE-PIC-Zn, which are similar in energy, tends to form the diastereopure crystal rather than the mixed one as explained above. They cannot form the RE-PIC-Zn·SE-PIC-Zn·2CHCl3 crystal due to the absence of CHCl3 molecules in spite of ∼11 kcal mol−1 preference. The CHCl3 solution of SE-PIC-Zn and RE-PIC-Zn, however, prefers to form the mixed crystal containing the CHCl3 molecules within the crystal rather than forming the diastereopure crystal. They do not form RE-PIC-Zn·SE-PIC-Zn because of its 18.8 kcal mol−1 higher energy.
Collet suggested that the preponderance of racemic crystals over conglomerates originates from the entropic free energy penalty of ca. 0.5 kcal mol−1 in conglomerates for demixing in the solid state.87,88 This level of energy difference was experimentally proved with metastable conglomerates and stable racemic crystals of 2-chloromandelic acid.89 Our results clearly indicated that the energy difference between diastereopure and diastereomeric mixture crystals is far beyond such a level and explains why the conversion into diastereomers from the racemate facilitates the enantioseparation. It is worth noting the energy difference between the two diastereomers, SE-PIC-Zn and RE-PIC-Zn, at this point. Both Ecoh and Ecryst of SE-PIC-Zn are commonly larger in magnitude than those of RE-PIC-Zn by 1.4 and 0.6 kcal mol−1, respectively. Although this level of energy difference between SE-PIC-Zn and RE-PIC-Zn is small relative to that between diastereopure and mixture crystals revealed in this study, it is comparable to that enough to segregate conglomerates and racemic crystals or that between polymorphs. The fact that the formation of the SE-PIC-Zn crystal precedes that of the RE-PIC-Zn one can be partly explained by this result.
4. Conclusion
In summary, we have developed a new enantioseparation method for PEA that consists of (i) combining (+)-camphor as a CDA by a condensation reaction, (ii) ZnCl2 coordination, (iii) separation of diastereomers by the solubility difference of diastereomers and fractional crystallization, (iv) demetallation, and (v) detaching the CDA by hydrolysis. The overall yields of both (S)-PEA and (R)-PEA were 24.8% with estimated ee of 98 and 99%, respectively. We have obtained the Ecoh of diastereopure crystals of both SE-PIC-Zn (−45.8 kcal mol−1) and RE-PIC-Zn (−44.4 kcal mol−1) as well as diastereomeric mixture crystals of RE-PIC-Zn·SE-PIC-Zn·2CHCl3 (−56.0 kcal mol−1) and RE-PIC-Zn·SE-PIC-Zn (−37.2 kcal mol−1) through DFT calculations under periodic boundary conditions with a plane wave basis set, which were based on the experimentally determined X-ray crystallographic data. We also obtained the Ecryst values through DFT calculations of the isolated molecule with a standard Gaussian basis set, which were −27.1 and −26.5 kcal mol−1 for SE-PIC-Zn and RE-PIC-Zn, respectively. These data provided the underlying background of spontaneous segregation of diastereopure crystals as well as preferred formation of the SE-PIC-Zn crystal over the RE-PIC-Zn one. This work suggested that the energy preference for forming diastereopure crystals over their corresponding mixture ones is ca. 8 kcal mol−1, which is far beyond the level of that discriminating conglomerates and racemic crystals (∼0.5 kcal mol−1). Our result gives a benchmark reference for future crystallization-based chiral resolution research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2017R1D1A3B03030670) and the National Research Foundation of Korea (NRF) grant funded by the Korea Government (2015R1D1A1A01058566 and 2018R1D1A1B07049976). This work was supported by the Korea Institute of Science and Technology Information with supercomputing resources including technical support (KSC-2016-C2-0049 and KSC-2017-C2-0040).References
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed.
- P. Cintas, Angew. Chem., Int. Ed., 2002, 41, 1139–1145 CrossRef CAS PubMed.
- A. M. Rouhi, Chem. Eng. News, 2003, 81, 56–61 CrossRef.
- L. Pasteur, C. R. Acad. Sci., 1848, 26, 535–539 Search PubMed.
- A. Collet, M. J. Brienne and J. Jacques, Chem. Rev., 1980, 80, 215–230 CrossRef CAS.
- W. H. Porter, Pure Appl. Chem., 1991, 63, 1119–1122 CAS.
- R. A. Sheldon, Chirotechnology, 1993, Mercel Dekker, N.Y. Search PubMed.
- A. Collet, Angew. Chem., Int. Ed., 1998, 37, 3239–3241 CrossRef CAS PubMed.
- R. Vespalec and P. Boček, Chem. Rev., 2000, 100, 3715–3753 CrossRef CAS PubMed.
- N. M. Maier, P. Franco and W. Lindner, J. Chromatogr. A, 2001, 906, 3–33 CrossRef CAS PubMed.
- C. A. M. Afonso and J. G. Crespo, Angew. Chem., Int. Ed., 2004, 43, 5293–5295 CrossRef CAS PubMed.
- N. G. Anderson, Org. Process Res. Dev., 2005, 9, 800–813 CrossRef CAS.
- T. Matsuura and H. Koshima, J. Photochem. Photobiol., C, 2005, 6, 7–24 CrossRef CAS.
- T. J. Ward, Anal. Chem., 2006, 78, 3947–3956 CrossRef CAS PubMed.
- B. Chankvetadze, J. Chromatogr. A, 2007, 1168, 45–70 CrossRef CAS PubMed.
- Z. Wang, J. Ouyang and W. R. G. Baeyens, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 862, 1–14 CrossRef CAS PubMed.
- I. Ilisz, R. Berkecz and A. Péter, J. Pharm. Biomed. Anal., 2008, 47, 1–15 CrossRef CAS PubMed.
- F. Faigl, E. Fogassy, M. Nógrádi, E. Pálovics and J. Schindler, Tetrahedron: Asymmetry, 2008, 19, 519–536 CrossRef CAS.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef CAS PubMed.
- M. Lämmerhofer, J. Chromatogr. A, 2010, 1217, 814–856 CrossRef PubMed.
- D. P. Glavin, J. E. Elsila, A. S. Burton, M. P. Callahan, J. P. Dworkin, R. W. Hilts and C. D. K. Herd, Meteorit. Planet. Sci., 2012, 47, 1347–1364 CrossRef CAS.
- D. Zbaida, M. Lahav, K. Drauz, G. Knaup and M. Kottenhahn, Tetrahedron, 2000, 56, 6645–6649 CrossRef CAS.
- M. P. Elsner, D.-F. Menéndez, E. A. Muslera and A. Seidel-Morgenstern, Chirality, 2005, 17, S183–S195 CrossRef CAS PubMed.
- G. Coquerel, Top. Curr. Chem., 2007, 269, 1–51 CAS.
- H. Lorenz, A. Perlberg, D. Sapoundjiev, M. P. Elsner and A. Seidel-Morgenstern, Chem. Eng. Process., 2006, 45, 863–873 CrossRef CAS.
- H. Lorenz, D. Polenske and A. Seidel-Morgenstern, Chirality, 2006, 18, 828–840 CrossRef CAS PubMed.
- N. Wermester, E. Aubin, M. Pauchet, S. Coste and G. Coquerel, Tetrahedron: Asymmetry, 2007, 18, 821–831 CrossRef CAS.
- M. P. Elsner, G. Ziomek and A. Seldel-Morgenstern, AIChE J., 2009, 55, 640–649 CrossRef CAS.
- D. G. Allen, G. M. McLaughlin, G. B. Robertson, W. L. Steffen, G. Salem and S. B. Wild, Inorg. Chem., 1982, 21, 1007–1014 CrossRef CAS.
- H. Lorenz, P. Sheehan and A. Seidel-Morgenstern, J. Chromatogr. A, 2001, 908, 201–214 CrossRef CAS PubMed.
- L. Mimassi, C. Guyard-Duhayon, M. N. Rager and H. Amouri, Inorg. Chem., 2004, 43, 6644–6649 CrossRef CAS PubMed.
- M. S. Kang, J. Cho, S. Nayab and J. H. Jeong, Polyhedron, 2019, 158, 135–143 CrossRef CAS.
- J. Cho, M. K. Chun, S. Nayab and J. H. Jeong, Transition Met. Chem., 2019, 44, 175–185 CrossRef CAS.
- S. Park, J. Lee, J. H. Jeong, H. Lee and S. Nayab, Polyhedron, 2018, 151, 82–89 CrossRef CAS.
- J. H. Ham, J. Cho, S. Nayab and J. H. Jeong, Inorg. Chim. Acta, 2018, 480, 33–41 CrossRef CAS.
- K. S. Kwon, S. Nayab and J. H. Jeong, Polyhedron, 2017, 130, 23–29 CrossRef CAS.
- G. Chelucci, S. Baldino, S. Chessa, G. A. Pinna and F. Soccolini, Tetrahedron: Asymmetry, 2006, 17, 3163–3169 CrossRef CAS.
- N. D. P. Cosford, L. Bleicher, A. Herbaut, J. S. McCallum, J.-M. Vernier, J. H. Dawson, J. P. Whitten, P. Adams, L. Chavez-Noriega, L. D. Correa, J. H. Crona, L. S. Mahaffy, F. Menzaghi, T. S. Rao, R. Reid, A. I. Sacaan, E. Santori, K. A. Stauderman, K. Whelan, G. K. Lloyd and I. A. McDonald, J. Med. Chem., 1996, 39, 3235–3237 CrossRef CAS PubMed.
- J.-L. Galzi and J.-P. Changeux, Neuropharmacology, 1995, 34, 563–582 CrossRef CAS.
- G. K. Lloyd and M. Williams, J. Pharmacol. Exp. Ther., 2000, 292, 461–467 CAS.
- F. F. Wagner and D. L. Comins, Org. Lett., 2006, 8, 3549–3552 CrossRef CAS PubMed.
- T. Tachibana, W. Ueoka, Md. S. I. Khan, R. Makino and M. A. Cline, Domest. Anim. Endocrinol., 2019, 66, 57–63 CrossRef CAS PubMed.
- E. Tamaddonfard, A. Erfanparast, H. Ghasemi, F. Henareh-Chareh, M. Hadidi, N. Mirzakhani, S. Seyedin, M. Taati, R. Salighedar, S. Salimi and F. Safaei, Eur. J. Pharmacol., 2016, 791, 696–702 CrossRef CAS PubMed.
- Y. T. Qin, S. H. Ma, Q. X. Zhuang, Y. H. Qiu, B. Li, Y. P. Peng and J. J. Wang, Neurosci. Lett., 2011, 502, 133–137 CrossRef CAS PubMed.
- H. E. Smith, L. J. Schaad, R. B. Banks, C. J. Wiant and C. F. Jordan, J. Am. Chem. Soc., 1973, 95, 811–818 CrossRef CAS.
- J. W. Canary, Y. Wang, R. Roy Jr., L. Que Jr. and H. Miyake, Inorg. Synth., 1998, 32, 70–75 CAS.
- J. W. Westley, R. H. Evans Jr. and J. F. Blount, J. Am. Chem. Soc., 1977, 99, 6057–6061 CrossRef CAS.
- L. E. Iglesias, V. M. Sánchez, F. Rebolledo and V. Gotor, Tetrahedron: Asymmetry, 1997, 8, 2675–2677 CrossRef CAS.
- K. A. Skupinska, E. J. McEachern, I. R. Baird, R. T. Skerlj and G. J. Bridger, J. Org. Chem., 2003, 68, 3546–3551 CrossRef CAS PubMed.
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Green Chem., 2017, 19, 474–480 RSC.
- J. Moellmann and S. Grimme, J. Phys. Chem. C, 2014, 118, 7615–7621 CrossRef CAS.
- A. Otero-de-la-Roza and E. R. Johnson, J. Chem. Phys., 2012, 137, 054103 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. e. a. Frisch, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- P. Fuentealba, H. Stoll, L. von Szentpaly, P. Schwerdtfeger and H. Preuss, J. Phys. B: At., Mol. Opt. Phys., 1983, 16, L323–L328 CrossRef CAS.
- U. Wedig, M. Dolg, H. Stoll and H. Preuss, Energy-Adjusted Pseudopotentials for Transition-Metal Elements, Springer, Dordrecht, 1986 Search PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Miertuš and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Angyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- G. Blay, E. Climent, I. Fernández, V. Hernández-Olmos and J. R. Pedro, Tetrahedron: Asymmetry, 2006, 17, 2046–2049 CrossRef CAS.
- G. Blay, E. Climent, I. Fernández, V. Hernández-Olmos and J. R. Pedro, Tetrahedron: Asymmetry, 2007, 18, 1603–1612 CrossRef CAS.
- S. Nayab and J. H. Jeong, Inorg. Chem. Commun., 2016, 65, 35–38 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- L. Kronik and A. Tkatchenko, Acc. Chem. Res., 2014, 47, 3208–3216 CrossRef CAS PubMed.
- K. Lejaeghere, G. Bihlmayer, T. Björkman, P. Blaha, S. Blügel, V. Blum, D. Caliste, I. E. Castelli, S. J. Clark, A. Dal Corso, S. De Gironcoli, T. Deutsch, J. K. Dewhurst, I. Di Marco, C. Draxl, M. Dułak, O. Eriksson, J. A. Flores-Livas, K. F. Garrity, L. Genovese, P. Giannozzi, M. Giantomassi, S. Goedecker, X. Gonze, O. Grånäs, E. K. U. Gross, A. Gulans, F. Gygi, D. R. Hamann, P. J. Hasnip, N. A. W. Holzwarth, D. Iuşan, D. B. Jochym, F. Jollet, D. Jones, G. Kresse, K. Koepernik, E. Küçükbenli, Y. O. Kvashnin, I. L. M. Locht, S. Lubeck, M. Marsman, N. Marzari, U. Nitzsche, L. Nordström, T. Ozaki, L. Paulatto, C. J. Pickard, W. Poelmans, M. I. J. Probert, K. Refson, M. Richter, G. M. Rignanese, S. Saha, M. Scheffler, M. Schlipf, K. Schwarz, S. Sharma, F. Tavazza, P. Thunström, A. Tkatchenko, M. Torrent, D. Vanderbilt, M. J. Van Setten, V. Van Speybroeck, J. M. Wills, J. R. Yates, G. X. Zhang and S. Cottenier, Science, 2016, 351, aad3000 CrossRef PubMed.
- B. M. Axilrod and E. Teller, J. Chem. Phys., 1943, 11, 299–300 CrossRef CAS.
- O. Anatole von Lilienfeld and A. Tkatchenko, J. Chem. Phys., 2010, 132, 234109 CrossRef PubMed.
- A. Otero-de-la-Roza and E. R. Johnson, J. Chem. Phys., 2013, 138, 054103 CrossRef CAS PubMed.
- T. Risthaus and S. Grimme, J. Chem. Theory Comput., 2013, 9, 1580–1591 CrossRef CAS PubMed.
- A. Collet, The homochiral versus heterochiral packing dilemma, in Chiral Molecules Problems and Wonders of Chiral Molecules, ed. M. Simonyi, Akad. Kiado, Budapest, Hung., 1990, pp. 91–109 Search PubMed.
- R. J. Davey, G. Sadiq, C. C. Seaton, R. G. Pritchard, G. Coquerel and C. Rougeot, CrystEngComm, 2014, 16, 4377–4381 RSC.
- H. Lorenz, J. von Langermann, G. Sadiq, C. C. Seaon, R. J. Davey and A. Seidel-Morgenstern, Cryst. Growth Des., 2011, 11, 1549–1556 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, 13C NMR spectra, IR spectra, DFT calculation results. CCDC 1914650–1914652. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi00523d |
| This journal is © the Partner Organisations 2019 |