
Enhanced tetragonality and large negative thermal expansion in a new Pb/Bi-based perovskite ferroelectric of (1 − x)PbTiO3–xBi(Zn1/2V1/2)O3†
Zhao
Pan
 *ab,
Jun
Chen
c,
Xingxing
Jiang
d,
Zheshuai
Lin
d,
Haibo
Zhang
a,
Yang
Ren
e,
Masaki
Azuma
*b and
Xianran
Xing
c
*ab,
Jun
Chen
c,
Xingxing
Jiang
d,
Zheshuai
Lin
d,
Haibo
Zhang
a,
Yang
Ren
e,
Masaki
Azuma
*b and
Xianran
Xing
c
aSchool of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: zhaopan@msl.titech.ac.jp
bLaboratory for Materials and Structures, Tokyo Institute of Technology, 4259 Nagatsuta, Midori, Yokohama, 226-8503, Japan. E-mail: mazuma@msl.titech.ac.jp
cDepartment of Physical Chemistry, University of Science and Technology Beijing, Beijing 100083, China
dInstitute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China
eX-Ray Science Division, Argonne National Laboratory, Argonne, Illinois 60439, USA
First published on 29th May 2019
Abstract
The exploration of a large negative thermal expansion (NTE) over a wide temperature range has been an important subject in materials science, since the overall coefficient of thermal expansion (CTE) of ordinary materials can be effectively tailored by the introduction of NTE materials. Here, we successfully achieved a large NTE within a broad temperature range in a new Pb/Bi-based ferroelectric of (1 − x)PbTiO3–xBi(Zn1/2V1/2)O3 by means of improving the ferroelectricity of PbTiO3. The present system exhibits an unusual enhanced tetragonality, large spontaneous polarization (PS), and high Curie temperature (TC). Specifically, the x = 0.1 compound exhibits an enhanced CTE of −2.10 × 10−5 °C−1 from room temperature (RT) up to its TC of 600 °C, which is in contrast to that of pristine PbTiO3 (−1.99 × 10−5 °C−1, RT−490 °C). More intriguingly, a large volume shrinkage (ΔV ≈ −1%) has also been observed during the ferroelectric-to-paraelectric phase transition. According to experimental and theoretical studies, the large NTE is attributed to the enhanced PS derived from the strong hybridization of Pb/Bi–O and Ti/Zn/V–O through the substitution of the polar Bi(Zn1/2V1/2)O3 perovskite. The present study demonstrates that a large NTE within a wide temperature range can be achieved in PbTiO3-based ferroelectrics by improving its ferroelectricity through introducing isostructural polar perovskites.
Introduction
Most materials display a positive thermal expansion (PTE) upon heating. However, although rare, several categories of materials exhibit a negative thermal expansion (NTE), in which the volume unusually contracts on heating. There are a number of important potential applications for materials with a negative coefficient of thermal expansion (CTE). Perhaps the most immediate one is in composite materials where the overall CTE can be precisely tailored to be a specific positive, negative or even zero value.1–6 The origins of NTE materials have been well studied in the past decades, mainly including the phonon-related transverse cooperative vibration (e.g., ZrW2O8, Ag3[Co(CN)6], and ScF3),7–10 the magnetovolume effect (e.g., Invar alloys and Mn3AN),11–14 the intermetallic charge transfer (e.g., LaCu3Fe4O12 and BiNiO3),15,16 the metal–insulator transition (e.g., CuRuO3),17,18 and the spontaneous volume ferroelectrostriction (SVFS) in PbTiO3-based ferroelectrics.19,20 Recent studies in the field of NTE materials have focused on a large NTE over a wide temperature range, due to the fact that normally the stronger the NTE is, the more efficient it is to counteract PTE within composite materials.2,4Among the available NTE materials, there is one branch of ferroelectrics which exhibit a general phenomenon in which the unit cell volume shrinks during the ferroelectric-to-paraelectric (FE–PE) phase transition, such as perovskites of PbTiO3,21–23 BaTiO3,24 (Bi, La)NiO3,16 and recently reported CH3NH3PbI3,25 tungsten bronze of PbNb2O6,26 and tin-hypothiodiphosphate of Sn2P2S6.27 However, all the ferroelectrics except PbTiO3, in which the NTE behavior occurs from room temperature to its Curie temperature (TC) of 490 °C, exhibit the NTE property in a relatively narrow temperature range.28 Such a wide NTE range of PbTiO3 provides the opportunity to modify the NTE property, i.e., achieving a large NTE over a wide temperature range. The origin of NTE in PbTiO3-based ferroelectrics has recently been well studied via the SVFS effect, which determines the crucial role of ferroelectric behavior in the NTE.19 Below TC, PbTiO3 exhibits ferroelectricity and NTE, while NTE disappears in the paraelectric phase above TC. In the ferroelectric phase, the increased volume can be well maintained by the large tetragonality (c/a) resulting from the strong spontaneous polarization (PS). It is worth noting that NTE in PbTiO3-based ferroelectrics is mainly attributed to shrinkage of the polar c axis. Based on these, it is therefore proposed that a large NTE could be obtained through improving the c/a of PbTiO3.
Bi(Zn1/2V1/2)O3 was recently reported to be a new PbTiO3-type perovskite, which exhibits a rather larger tetragonality (c/a = 1.26) and stronger polarization (PS = 126 μC cm−2) compared to those of PbTiO3 (c/a = 1.064, PS = 59 μC cm−2).29,30 The introduction of Bi(Zn1/2V1/2)O3 in PbTiO3 is considered to further increase its tetragonality, and therefore achieve an enhanced NTE. Herein, we successfully achieved a large NTE over a wide temperature range in the present new Pb/Bi-based binary system of (1 − x)PbTiO3–xBi(Zn1/2V1/2)O3. A continuously enhanced c/a has been observed by the substitution of the polar Bi(Zn1/2V1/2)O3 perovskite. As expected, both the NTE and TC have been improved to a certain extent. In particular, remarkably volume shrinkages have also been observed in (1 − x)PbTiO3–xBi(Zn1/2V1/2)O3 during the FE–PE phase transition, which have been well studied by the experimental and theoretical results.
Experimental section
Material synthesis and measurements
Polycrystalline samples of (1 − x)PbTiO3–xBi(Zn1/2V1/2)O3 (hereafter abbreviated as (1 − x)PT–xBZV, x = 0.1–0.5) were prepared by using a cubic anvil-type high-pressure apparatus. The raw materials of PbO, Bi2O3, ZnO, TiO2, V2O3, and V2O5 were stoichimetrically mixed and sealed in a gold capsule, and then treated at 6 GPa and 1100 °C for 30 min. After the high-pressure process, the obtained compounds were carefully ground in an agate mortar, and subsequently annealed at 400 °C for 4 hours. The X-ray diffraction (XRD) patterns were collected with a Bruker D8 AVANCE diffractometer for phase identification. The high-temperature synchrotron powder diffraction (SXRD) experiment was conducted at the 11-ID-C beamline of the Advanced Photon Source with a light wavelength of 0.117418 Å.Computational
First-principles calculation was performed by using CASTEP,31 a total energy package based on the plane-wave pseudopotential density functional theory (DFT).32,33 The correlation and exchange energy was described by Perdew, Burke and Ernzerhof (PBE)34 in the generalized gradient approximation (GGA)35 form. The optimized norm-conserving pseudopotential36 in the Kleinman–Bylander37 form was adopted to model the effective interaction between the valence electrons and atom cores, allowing the choice of a relatively small plane-wave basis set without compromising the computational accuracy. The disorder occupation in 0.5PbTiO3–0.5Bi(Zn1/2V1/2)O3 was handled by virtual crystal approximation (VCA),38 in which the disorder position is modeled by a ghost atom with the effective Coulomb potential weighted by its atomic constitution. A kinetic energy cutoff of 800 eV and a Monkhorst–Pack39 with k-point meshes less than 0.03 Å−1 in the Brillouin zones were chosen.Results and discussion
The laboratory XRD patterns of (1 − x)PT–xBZV from x = 0.1 to 0.5 are shown in Fig. 1(a). As can be seen, all the investigated compounds exhibit a single tetragonal phase without any noticeable impurities. With the substitution of BZV, the (001) peak shows a clear shift to the lower angle, indicating the expansion of the c axis. While the (100) peak exhibits an opposite trend, it shifts slightly to the higher angle which demonstrates the shrinkage of the a(b) axis. The detailed lattice parameters were refined and plotted in Fig. 1(b). The c axis increases almost linearly, whereas the a(b) axis decreases continuously as a function of BZV. Consequently, an unusual enhanced c/a has been observed. The c/a value increases from 1.06 of pristine PT to 1.08, 1.12, 1.13, and 1.16 of x = 0.1, 0.2, 0.3, 0.4, and 0.5, respectively, which suggests that the substitution of BZV effectively enhanced the tetragonality of PT. Here, the large tetragonality results in a pyramidal rather than an octahedral coordination in the (1 − x)PT–xBZV solid solutions (see the top left of Fig. 2). The large lattice distortion can be attributed to the large PS displacements induced by the strong Pb/Bi–O hybridization and coupling interactions between Ti/Zn/V and Pb/Bi cations, which can be evidenced by the following theoretical calculations.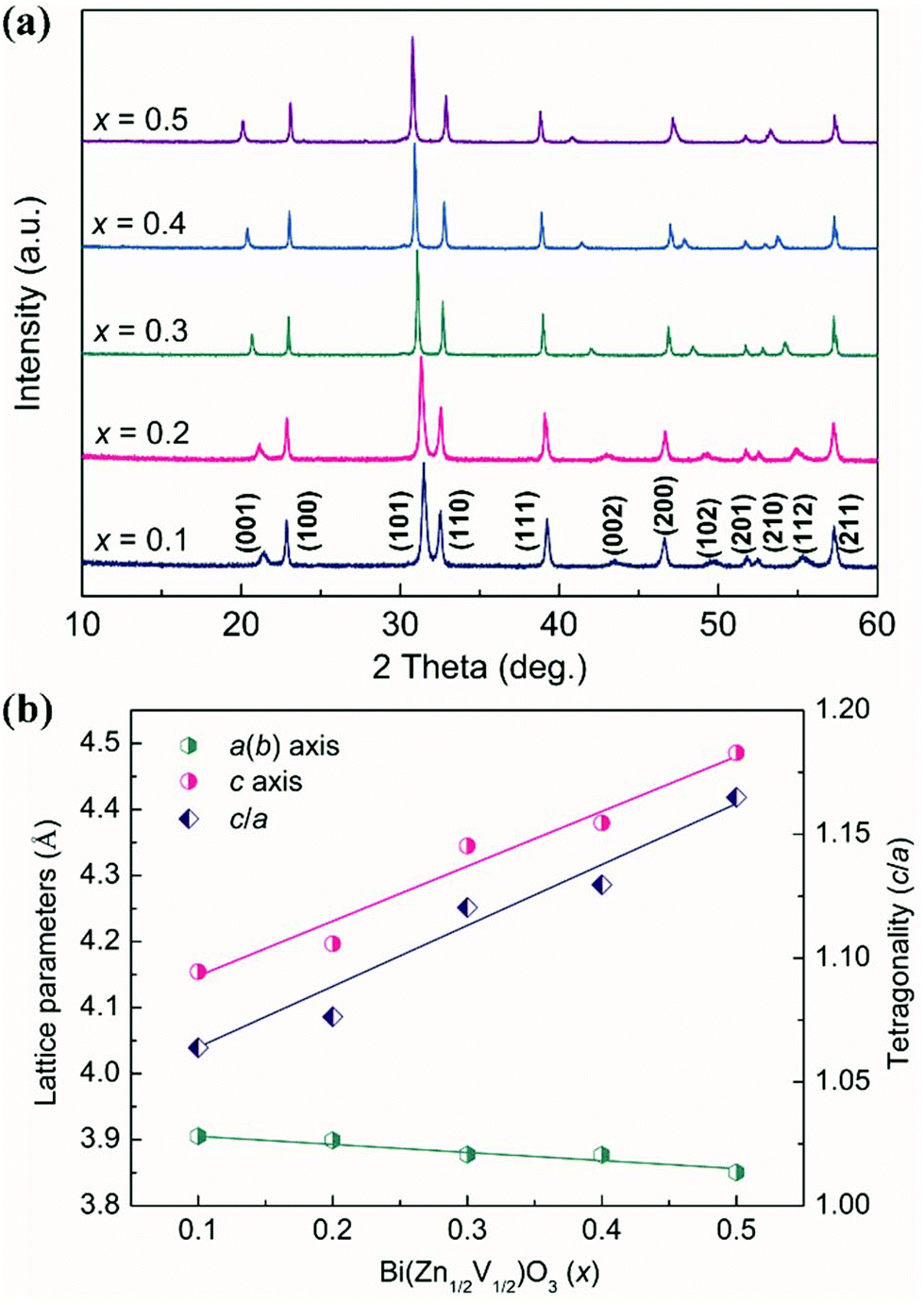 | ||
| Fig. 1 (a) XRD patterns and (b) lattice parameters of (1 − x)PT–xBZV (x = 0.1–0.5) at room temperature. | ||
 | ||
| Fig. 2 The calculated spontaneous polarization of (1 − x)PT–xBZV (x = 0.1–0.5) at room temperature. The insets are the crystal structure (top left) and schematic diagram of PS (bottom right). | ||
It is well known that in ABO3 perovskite-type ferroelectrics, PS originates from the displacements from the centroid of the oxygen polyhedrons of the A site and B site atoms (see the bottom right of Fig. 2).21 Here, the PS displacements of the A site Pb/Bi and the B site Ti/Zn/V cations can be derived from the SXRD refinement results. The detailed structure of (1 − x)PT–xBZV was refined using the Rietveld method with the FullProf software. The initial structural model corresponds to PbTiO3 with the atomic positions being given in the noncentrosymmetric space group P4mm (no. 99).40 The occupancies of atoms were fixed at the ideal composition. The isotropic thermal factors of Pb/Bi, Zn/Ti/V, as well as O(I) and O(II) were set equal, respectively. In addition, the anisotropic profile broadening model has been adopted in order to obtain good fitting results.41 As can be seen, the calculated diffraction profiles agree well with the observed ones (Fig. S1–S5†). The related PS can be estimated by considering a purely ionic crystal and neglecting the electronic polarization by using the following formula,42

Both theoretical and experimental studies have confirmed that enhanced tetragonality in the PbTiO3-based ferroelectric could give rise to an enhanced NTE.19,43 Generally, a large c/a indicates a large lattice distortion, and could result in large volume shrinkage with the release of high lattice energy during the heating process. As a result, an enhanced NTE could be observed, such as in Pb1−xCdxTiO3,44 PbTiO3–BiFeO3,45 and recently reported Pb(Ti1−xVx)O3 and (1 − x)PbTiO3–xBiCoO3 systems,20,46 which exhibit abnormally enhanced c/a and NTE compared to those of PbTiO3. In order to precisely investigate the thermal expansion property of the (1 − x)PT–xBZV system, the temperature dependence of SXRD experiments was determined (Fig. S6 and S7†). The detailed unit cell volumes were extracted by means of structure refinement based on the SXRD data (Fig. 3a). With the substitution of BZV, the 0.9PT–0.1BZV compound presents a nonlinear and strong NTE in a wide temperature range from room temperature (RT) to near TC. This is in contrast to pristine BZV, which decomposes at a temperature higher than 730 K before reaching its TC.29 Note that the unit cell volume of 0.9PT–0.1BZV shows little dependence on temperature before the FE–PE phase transition. However, it contracts dramatically during the FE–PE phase transition. A noticeable volume shrinkage of −0.95% has been observed during the phase transition. The average CTE of the overall temperature range is −2.10 × 10−5 °C−1 (RT ∼ 600 °C). With further increase of the content of BZV, the compound of 0.8PT–0.2BZV shows a tendency of PTE with a negligible CTE of 1.56 × 10−6 °C−1 (RT ∼ 680 °C) before the FE–PE phase transition. Intriguingly, a more pronounced volume contraction as large as −1.08% has been observed during the FE–PE phase transition. In comparison, the present NTE is among giant NTE materials such as CuO nanoparticles (−1.1%),47 Mn3AN (−1.3%),12 BiNiO3 (−2.5%),16 Ca2RuO3.74 (−1.0%),18 and recently reported Pb0.76La0.04Bi0.20VO3 (−6.7%).48 It is worth noting that the present compound of 0.9PT–0.1BZV (−2.10 × 10−5 °C−1, RT ∼ 600 °C) not only enhances the NTE of PT (−1.99 × 10−5 °C−1, RT ∼ 490 °C) but also extends the NTE to a much wider temperature range. The TC of PT has been significantly increased by 100 °C, which is attributed to the enhanced polarization according to the empirical relationship of TC = αPS2.30 For further increasing the content of BZV, the compounds decompose before reaching the FE–PE phase transition temperature, which is ascribed to the increased lattice distortion and weakened thermal stability of the perovskite structure.
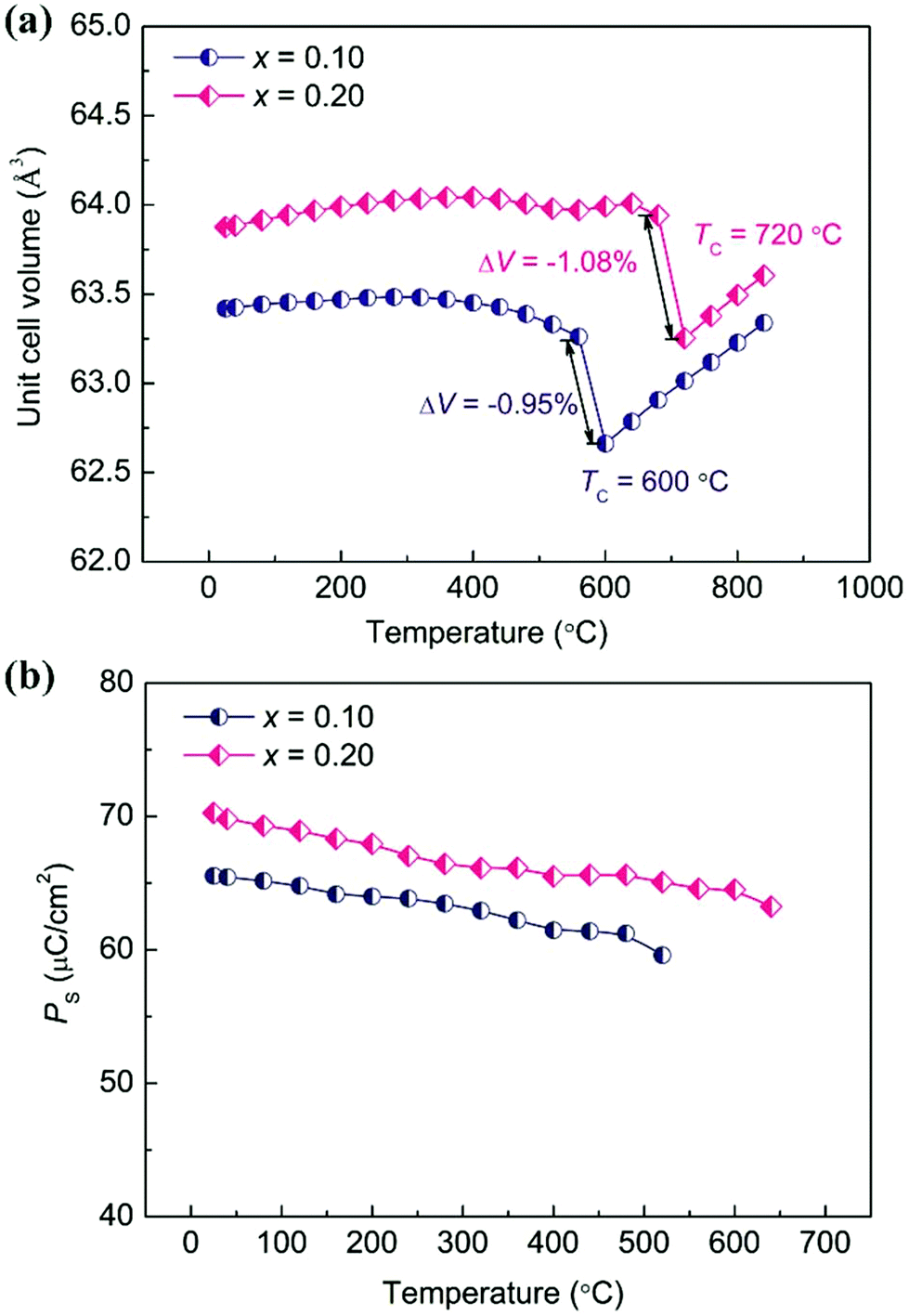 | ||
| Fig. 3 (a) Unit cell volume and (b) calculated spontaneous polarization of (1 − x)PT–xBZV (x = 0.1 and 0.2) as a function of temperature. | ||
Recently, a new concept of spontaneous volume ferroelectrostriction (SVFS) has been proposed to illuminate the origin of NTE in PbTiO3-based ferroelectrics.19 According to the description of SVFS, the ferroelectric behavior plays an important role in the NTE of PT-based ferroelectrics. The NTE of PT-based ferroelectrics generally occurs in the ferroelectric phase, while it disappears in the paraelectric phase. The increased volume in the ferroelectric phase can be maintained by the high lattice distortion c/a which results from the large PS. Therefore, how the PS, which can reflect the magnitude of ferroelectricity, varies with temperature, will directly affect the NTE. The variation of PS as a function of temperature for 0.9PT–0.1BZV and 0.8PT–0.2BZV is shown in Fig. 3b. The PS values of both the two compounds exhibit a slightly decreasing tendency with increasing temperature, which is ascribed to the weakened ferroelectricity. As the temperature is raised up to near TC, PS still remains at a high level, with the values of 60 and 65 μC cm−2 for 0.9PT–0.1BZV and 0.8PT–0.2BZV, respectively. Correspondingly, the unit cell volumes of 0.9PT–0.1BZV and 0.8PT–0.2BZV can be retained close to that of room temperature by the high PS. As a result, both the two compositions exhibit little temperature dependence of unit cell volumes before reaching their TCs. However, PS suddenly disappears on approaching the TC, resulting in noticeable volume shrinkages during the FE-to-PE phase transition.
To further study the effect of BZV substitution on the hybridization with oxygen atoms, the electron density difference of (1 − x)PT–xBZV (x = 0 and 0.5) was calculated by first-principles calculation. As depicted in Fig. 4, it is revealed that in both PbTiO3 and 0.5PbTiO3–0.5Bi(Zn0.5V0.5)O3, the concentration of the valence electrons on the Ti–O (or Ti0.5Zn0.25V0.25–O) bond along the c-axis is more prominent than that within the (a,b) plane, which indicates that the orbital hybridization along the c-axis is stronger than that along the a(b)-axis. In pristine PbTiO3, apart from the electronic cloud concentration, some electron-loss area also occurs, while in 0.5PbTiO3–0.5BiZn0.5V0.5O3, the electron concentration strongly dominates in the chemical bonds within the pyramidal anions. Moreover, the highest value of the electronic density difference in PbTiO3 is 3.75 Å−3, which is smaller than that in 0.5PbTiO3–0.5BiZn0.5V0.5O3 (4.75 Å−3). These observations demonstrate that the Zn0.5V0.5/Ti substitution strongly enhanced the covalent interaction within Ti0.5Zn0.25V0.25O3 pyramid, giving rise to the larger spontaneous polarization.
 | ||
| Fig. 4 The electronic density difference map of (a) PbTiO3 and (b) 0.5PT–0.5BZV. The Pb(Pb/Bi), Ti(Ti/Zn/V), and O atoms are indicated by black, blue, green, and violet balls, respectively. | ||
Conclusions
In summary, a new Pb/Bi-based ferroelectric based on (1 − x)PT–xBZV has been designed to exhibit a large NTE over a wide temperature range. Both the tetragonality and Curie temperature are considerably increased. The 0.9PT–0.1BZV compound exhibits a large NTE with an average CTE of −2.10 × 10−5 °C−1 in the temperature range from room temperature up to 600 °C, which is largely enhanced compared to that of PbTiO3. In particular, large volume shrinkages have also been observed in 0.9PT–0.1BZV and 0.8PT–0.2BZV during the FE–PE phase transition. The large NTE is closely related to the enhanced PS due to the substitution of BZV, which has been well elaborated by the experimental and theoretical results. The present study demonstrates that a large NTE within the extended temperature range could be achieved in PbTiO3-based ferroelectrics through improving its ferroelectric property.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21805215), the National Program for Support of Top-notch Young Professionals, the Program for Changjiang Young Scholars, and the General Financial Grant from the China Postdoctoral Science Foundation (2017M622536). The use of the Advanced Photon Source at the Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Science (DE-AC02-06CH11357).Notes and references
- J. S. O. Evans, J. Chem. Soc., Dalton Trans., 1999, 19, 3317–3326 RSC.
- K. Takenaka, Sci. Technol. Adv. Mater., 2012, 13, 013001 CrossRef.
- M. Azuma, K. Oka and K. Nabetani, Sci. Technol. Adv. Mater., 2015, 16, 034904 CrossRef PubMed.
- J. Chen, L. Hu, J. X. Deng and X. R. Xing, Chem. Soc. Rev., 2015, 44, 3522–3567 RSC.
- J. P. Attfield, Front. Chem., 2018, 6, 371 CrossRef PubMed.
- X. Jiang, Y. Yang, M. S. Molokeev, P. F. Gong, F. Liang, S. H. Wang, L. Liu, X. Wu, X. D. Li, Y. C. Li, S. F. Wu, W. Li, Y. C. Wu and Z. S. Lin, Adv. Mater., 2018, 30, 1801313 CrossRef PubMed.
- T. A. Mary, J. S. O. Evans, T. Vogt and A. W. Sleight, Science, 1996, 272, 90–92 CrossRef CAS.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- B. K. Greve, K. L. Martin, P. L. Lee, P. J. Chupas, K. W. Chapman and A. P. Wilkinsom, J. Am. Chem. Soc., 2010, 132, 15496–15498 CrossRef CAS PubMed.
- L. Hu, J. Chen, A. Sanson, H. Wu, C. G. Rodriguez, L. Olivi, Y. Ren, L. L. Fan, J. X. Deng and X. R. Xing, J. Am. Chem. Soc., 2016, 138, 8320–8323 CrossRef CAS PubMed.
- M. Van Schilfgaarde, I. A. Abrikosov and B. Johansson, Nature, 1999, 400, 46–49 CrossRef CAS.
- K. Takenaka and H. Takagi, Appl. Phys. Lett., 2005, 87, 261902 CrossRef.
- Y. Y. Zhao, F. X. Hu, L. F. Bao, J. Wang, H. Wu, Q. Z. Huang, R. R. Wu, Y. Liu, F. R. Shen, H. Kuang, M. Zhang, W. L. Zuo, X. Q. Zheng, J. R. Sun and B. G. Shen, J. Am. Chem. Soc., 2015, 137, 1746–1749 CrossRef CAS.
- Y. Sun, C. Wang, Y. C. Wen, L. H. Chu, H. Pan and M. Nie, J. Am. Ceram. Soc., 2010, 93, 2178–2181 CrossRef CAS.
- Y. W. Long, N. Hayashi, T. Saito, M. Azuma, S. Muranaka and Y. Shimakawa, Nature, 2009, 458, 60–63 CrossRef CAS PubMed.
- M. Azuma, W. T. Chen, H. Seki, M. Czapski, S. Olga, K. Oka, M. Mizumaki, T. Watanuki, N. Ishimatsu, N. Kawamura, S. Ishiwata, M. G. Tucker, Y. Shimakawa and J. P. Attfield, Nat. Commun., 2011, 2, 347 CrossRef PubMed.
- T. F. Qi, O. B. Korneta, S. Parkin, L. E. De Long, P. Schlottmann and G. Cao, Phys. Rev. Lett., 2010, 105, 177203 CrossRef CAS PubMed.
- K. Takenaka, Y. Okamato, T. Shinoda, N. Katayama and Y. Sakai, Nat. Commun., 2017, 8, 14102 CrossRef CAS PubMed.
- J. Chen, F. F. Wang, Q. Z. Huang, L. Hu, X. P. Song, J. X. Deng, R. B. Yu and X. R. Xing, Sci. Rep., 2013, 3, 2458 CrossRef PubMed.
- Z. Pan, J. Chen, X. X. Jiang, L. Hu, R. Z. Yu, H. Yamamoto, T. Ogata, Y. Hattori, F. M. Guo, X. A. Fan, Y. W. Li, G. Q. Li, H. Z. Gu, Y. Ren, Z. S. Lin, M. Azuma and X. R. Xing, J. Am. Chem. Soc., 2017, 139, 14865–14868 CrossRef CAS PubMed.
- J. Chen, X. R. Xing, C. Sun, P. H. Hu, R. B. Yu, X. W. Wang and L. H. Li, J. Am. Chem. Soc., 2008, 130, 1144–1145 CrossRef CAS PubMed.
- P. H. Hu, H. J. Kang, J. Chen, J. X. Deng and X. R. Xing, J. Mater. Chem., 2011, 21, 16205–16209 RSC.
- Z. Pan, J. Chen, X. Jiang, Z. S. Lin, L. L. Fan, Y. C. Rong, L. Hu, H. Liu, Y. Ren, X. J. Kuang and X. R. Xing, Inorg. Chem., 2017, 56, 2589–2595 CrossRef CAS PubMed.
- G. Shirane and A. Takeda, J. Phys. Soc. Jpn., 1952, 7, 1–4 CrossRef CAS.
- P. A. Mante, C. C. Stoumpos, M. G. Kanatzidis and A. Yartsev, J. Phys. Chem. Lett., 2018, 9, 3161–3166 CrossRef CAS PubMed.
- E. C. Subbarao, J. Am. Ceram. Soc., 1960, 4, 439–442 CrossRef.
- Y. C. Rong, M. L. Li, J. Chen, M. Zhou, K. Lin, L. Hu, W. X. Yuan, W. H. Duan, J. X. Deng and X. R. Xing, Phys. Chem. Chem. Phys., 2016, 18, 6247–6251 RSC.
- X. R. Xing, J. X. Deng, J. Chen and G. R. Liu, Rare Met., 2003, 22, 294–297 CAS.
- R. Yu, H. Hojo, K. Oka, T. Watanuki, A. Machida, K. Shimizu, K. Nakano and M. Azuma, Chem. Mater., 2015, 27, 2012–2017 CrossRef CAS.
- S. C. Abrahams, S. K. Kurtz and P. B. Jamieson, Phys. Rev., 1968, 172, 551–553 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr.–Cryst. Mater., 2005, 220, 567–570 CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 12947–12954 CrossRef.
- D. R. Hamann, M. Schluter and C. Chiang, Phys. Rev. Lett., 1979, 43, 1494–1497 CrossRef CAS.
- L. Kleinman and D. M. Bylander, Phys. Rev. Lett., 1982, 48, 1425–1428 CrossRef CAS.
- L. Bellaiche and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 7877–7882 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- G. Shirane, R. Pepinsky and B. C. Frazer, Acta Crystallogr., 1956, 9, 131–140 CrossRef CAS.
- P. W. Stephens, J. Appl. Crystallogr., 1999, 32, 281–289 CrossRef CAS.
- J. Frantti, S. Ivanov, S. Eriksson, H. Rundlöf, V. Lantto, J. Lappalainen and M. Kakihana, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 064108 CrossRef.
- F. F. Wang, Y. Xie, J. Chen, H. G. Fu and X. R. Xing, Phys. Chem. Chem. Phys., 2014, 16, 5237–5241 RSC.
- J. Chen, X. R. Xing, R. B. Yu and G. R. Liu, Appl. Phys. Lett., 2005, 87, 231915 CrossRef.
- J. Chen, X. R. Xing, G. R. Liu, J. H. Li and Y. T. Liu, Appl. Phys. Lett., 2006, 89, 101914 CrossRef.
- Z. Pan, J. Chen, R. Z. Yu, L. Patra, P. Ravindran, A. Sanson, R. Milazzo, A. Carnera, L. Hu, L. Wang, H. Yamamoto, Y. Ren, Q. Z. Huang, Y. Sakai, T. Nishikubo, T. Ogata, X. A. Fan, Y. W. Li, G. Q. Li, H. Hojo, M. Azuma and X. R. Xing, Chem. Mater., 2019, 31, 1296–1303 CrossRef CAS.
- X. G. Zheng, H. Kubozono, H. Yamada, K. Kato, Y. Ishiwata and C. N. Xu, Nat. Nanotechnol., 2008, 3, 724–726 CrossRef CAS PubMed.
- H. Yamamoto, T. Imai, Y. Sakai and M. Azuma, Angew. Chem., Int. Ed., 2018, 57, 8170–8173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qi00450e |
| This journal is © the Partner Organisations 2019 |