DOI:
10.1039/C9QI00356H
(Research Article)
Inorg. Chem. Front., 2019,
6, 1861-1872
In Operando analysis of the charge storage mechanism in a conversion ZnCo2O4 anode and the application in flexible Li-ion batteries†
Received
29th March 2019
, Accepted 27th May 2019
First published on 28th May 2019
Abstract
As a conversion-type electrode material, ZnCo2O4 (ZCO) is intensively researched due to its attractive high specific capacity. Much effort to study ZCO supported on a conductive matrix has been successful to overcome the inherent drawbacks of low conductivity and dramatic volume variation during the (de)lithiation process. Despite many reported studies, the lithiation storage mechanism in the ZCO electrode is not yet clearly elucidated. In this work, in operando synchrotron radiation diffraction and in operando X-ray absorption spectroscopy are used to study the lithium storage mechanism in the ZCO material. The initial conversion process of ZnCo2O4, involving multiple reactions based on intercalation, conversion and alloying is deeply elucidated. During the 1st lithiation intermediate phases such as LiCo2O3, CoO and ZnO are formed. On the other hand, upon delithiation, the conversion to ZnO and CoO (and not to the pristine ZnCo2O4) occurs. This is different from the previous conclusion, which claims that Co3O4 forms after the initial delithiation. Furthermore, a binder-free ZnCo2O4/carbon cloth composite electrode is also prepared, which exhibits higher rate performance and capacity retention, compared to the bare ZCO electrode.
Introduction
Nowadays, Li-ion batteries (LIBs) with long lifespan and high energy density have attracted much attention to meet the increasing demand of electric devices and electric vehicles in our daily life.1,2 Nevertheless, the low capacity of commercial graphite (theoretical capacity = 372 mA h g−1) is a major challenge to overcome.3 Therefore, the interest in conversion-type transition metal oxide anode materials increases due to their high specific capacity, which is 2–3 times higher than that of graphite.4,5 In addition, high reaction potentials can reduce the possibility of lithium dendrite formation, improving safety performance.6–8 However, some intrinsic problems such as low conductivity and particle pulverization are harmful for rate capability and cycling life.9 Therefore, an interesting strategy, transition metal oxide loading on a conductive matrix, was proposed, because this design can not only increase the electronic conductivity but also relieve repeated volume change.10,11
So far, many transition metal oxides have been intensively studied as anode materials for LIBs.12 As it is known, cobalt oxides have high structural and cycling stability, compared to other transition metal oxides.13 However, the high-cost and high-toxicity of cobalt block the large-scale application of cobalt oxides. One strategy to reduce the usage of cobalt is its partial substitution with inexpensive and nontoxic elements. In this line, some ternary oxides have been developed, such as ZnCo2O4,13–15 CoFe2O4,16–18 and NiCo2O4.19–21 Among them, ZnCo2O4 (ZCO) is very promising since conversion and alloy combined charge storage mechanism occurs upon lithiation. This material delivers high specific capacity, theoretically 789 mA h g−1 with 7e− per formula unit (pfu).22,23 Furthermore, some hierarchical ZCO structures loaded on a conductive matrix were designed to alleviate volume change, increase electron conductivity and shorten the Li+ pathway, such as peanut-like ZCO/rGO/CNTs,24 ZCO-urchins/carbon-fibers,25 ZCO nanowire arrays/carbon cloth,22 ZCO nanosheets on nickel foam,26 and ZCO nanoflowers/carbon nanotube/N-doped graphene.27
Despite the widely available literature related to the synthesis of ZCO and hierarchical ZCO/matrix composites and their improved electrochemical performance, there is still no deep understanding of the working mechanism of ZCO. Up to now, ex situ X-ray diffraction (XRD), transmission electron microscopy (TEM), and selected area electron diffraction (SAED) have been employed as tools for elucidating the conversion mechanism, revealing the formation of LiZn ↔ Zn ↔ ZnO and Co ↔ CoO ↔ Co3O4 during cycling.5 Some reports involving other transition metal oxides, such as ZnO/ZnFe2O428 and ZnMn2O4,29 clearly demonstrate the phase evolution during the first cycle via in situ XRD method. Therefore, in operando techniques are powerful methods to establish and understand the mechanism of (de)lithiation. However, it is difficult to do this directly on a hierarchical ZCO/matrix because of the low ZCO loading. Very recently, Deng et al. used in situ XRD to study the lithium storage in ZCO spheres, but they only observed gradual fading of characteristic diffraction peaks of ZCO upon the initial lithiation.15 Because the initial lithiation process is crucial to electrochemically activate the ZCO material, it is necessary to investigate this material in-depth via in operando techniques.
Inspired by the above discussion, this work aims to elucidate the Li storage mechanism in hierarchical ZCO/carbon cloth (ZCO/CC) composites, and parallelepipedal ZCO particles via in operando synchrotron radiation diffraction (SRD) and X-ray absorption spectroscopy (XAS). Notably, the ZCO and the ZCO/CC were synthesized in the same autoclave under the same synthesis conditions, to ensure the same crystal properties. This study reveals the real potentials when the intermediate phases form and vanish during the conversion process. Afterwards, the hierarchical ZCO/CC composite was used as binder-free anodes for lithium storage. The electrochemical performance of the ZCO/CC and bare ZCO electrodes was measured and compared.
Experimental section
Simultaneous synthesis of ZCO and ZCO/CC
Following the schematic diagram depicted in Fig. 1, ZnSO4·6H2O (2 mmol, 99.0%, Acros), Co(NO3)2·6H2O (4 mmol, 98.0%, Sigma-Aldrich) and urea (15 mmol, 98.0%, Sigma-Aldrich) were dissolved in 70 mL deionized water by magnetic stirring for 30 min. Then, the CC (25 mm × 25 mm, H2315/H23, Quin Tech) was vertically fixed in the center of a 100 mL Teflon-lined autoclave. The mixed solution was later put into the same autoclave and heated at 200 °C for 16 h. After cooling down, the two precursors (the ZCO particle sediment and the ZCO/CC composite) were obtained simultaneously in one pot. After that, the two obtained precursors were washed separately with deionized water and absolute ethanol several times and then dried at 80 °C overnight. Finally, the ZCO precursor was sintered at 500 °C for 6 h in air, whereas the ZCO/CC precursor was annealed at 500 °C for 6 h under N2 flow (in order to avoid the combustion of the CC) and afterwards sintered at 450 °C for 15 min in air for further oxidation of Co2+ to Co3+. A heating rate of 5 °C min−1 was used until the annealing temperature was reached.
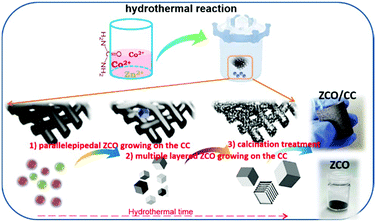 |
| | Fig. 1 Schematic diagram of the hydrothermal synthesis process of the ZCO and the ZCO/CC. | |
Electrochemical characterization
ZCO electrodes were prepared by mixing ZCO powder, carbon black (Super-C65, Timcal Ltd) and PVDF binder (R6020/1001, Solvay) in a weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 using NMP (GC 99.5%, Merck KGaA). The slurry was cast on Cu foil using an Erichsen coatmaster with a doctor-blade. After drying, the coated foil was cut into disks (∅ 12 mm, ∼14 μm thick and ∼1.1 mg ZCO loading on ∼10 mg Cu foil). As a comparison, the as-prepared ZCO/CC composite was also cut into disks (∅ 12 mm, ∼147 μm thick, and ∼1.9 mg ZCO loading on 4.5 mg carbon cloth). The working electrode, polypropylene separator (∅ 17 mm, Celgard 2325-1750-A), lithium foil counter electrode (∅ 15 mm, Alfa Aesar) and LP30 electrolyte (1 M LiPF6 in ethylene carbonate/dimethyl carbonate = 1:1 in mass, BASF) were used to build CR2032 coin cells in an argon-filled glovebox (MB200, MBraun GmbH). Galvanostatic cycling (GC), cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were carried out utilizing a multichannel potentiostat (VMP3, Bio-Logic). The cells were kept in a Binder climate chamber at 25 °C during the electrochemical experiments. GC and CV were conducted in a potential range of 3.0–0.01 V vs. Li/Li+. EIS measurements were conducted at 3.0 V upon an alternating current signal with an amplitude of 10 mV in a frequency range from 100 kHz to 10 mHz.
:1:1 using NMP (GC 99.5%, Merck KGaA). The slurry was cast on Cu foil using an Erichsen coatmaster with a doctor-blade. After drying, the coated foil was cut into disks (∅ 12 mm, ∼14 μm thick and ∼1.1 mg ZCO loading on ∼10 mg Cu foil). As a comparison, the as-prepared ZCO/CC composite was also cut into disks (∅ 12 mm, ∼147 μm thick, and ∼1.9 mg ZCO loading on 4.5 mg carbon cloth). The working electrode, polypropylene separator (∅ 17 mm, Celgard 2325-1750-A), lithium foil counter electrode (∅ 15 mm, Alfa Aesar) and LP30 electrolyte (1 M LiPF6 in ethylene carbonate/dimethyl carbonate = 1:1 in mass, BASF) were used to build CR2032 coin cells in an argon-filled glovebox (MB200, MBraun GmbH). Galvanostatic cycling (GC), cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were carried out utilizing a multichannel potentiostat (VMP3, Bio-Logic). The cells were kept in a Binder climate chamber at 25 °C during the electrochemical experiments. GC and CV were conducted in a potential range of 3.0–0.01 V vs. Li/Li+. EIS measurements were conducted at 3.0 V upon an alternating current signal with an amplitude of 10 mV in a frequency range from 100 kHz to 10 mHz.
General characterization
XRD was carried out using STOE STADI P X-ray powder diffractometers equipped with Mythen1K detectors. Mo Kα1 radiation (λ = 0.70932 Å) and Co Kα radiation (λ = 1.78896 Å) were used to characterize the crystal structure. The morphology of the samples was investigated by using a scanning electron microscope (SEM, Merlin, Zeiss GmbH). The elemental distribution of the samples was determined by energy-dispersive X-ray spectroscopy (EDS) with a Bruker XFlash (60 mm2) EDS detector. Thermogravimetry (TG) data were collected through an STA 449C Netzsch analyzer from 35 °C to 1000 °C with a heating rate of 5 °C min−1 under oxygen/argon flow (11/31 mL min−1). Raman measurements were performed by using a Raman spectrometer (LabRam Evolution HR, Horiba Jobin Yvon) using 532 nm laser excitation with a power of 10 mW. X-ray photoelectron spectroscopy (XPS) was performed using a K-Alpha+XPS spectrometer (ThermoFisher Scientific, East Grinstead, UK). The ZCO powder was analyzed using a microfocused, monochromated Al Kα X-ray source (400 μm spot size). The K-Alpha+charge compensation system was employed during analysis, using electrons of 8 eV energy and low-energy argon ions to prevent any localized charge build-up. The spectra were fit with one or more Voigt profiles (binding energy uncertainty: ±0.2 eV) and Scofield sensitivity factors were applied for quantification.30 All spectra were referenced to the C 1s peak (C–C, C–H) at 285.0 eV binding energy controlled by means of the photoelectron peaks of metallic Cu, Ag, and Au, respectively.
In operando characterization
The electrodes for in operando SRD and XAS were prepared by mixing 70 wt% of the ZCO active material, 20 wt% of carbon black (Super P Li, Timcal Ltd) and 10 wt% of polytetrafluoroethylene (PTFE, white beads, Aldrich) binder by dry grinding and pressing on Cu mesh. About 5 mg of the mixture was pressed on the center of a copper mesh (∅ 12 mm) and dried in a vacuum at 80 °C for 24 h. Li|LP30|ZCO in operando coin cells (CR2025 with a window in the center) were assembled by the same method stated above. The cells were cycled with a static current of 75 mA g−1 in a potential range of 0.01–3.0 V vs. Li/Li+ at room temperature.
In operando XRD data were collected by using synchrotron radiation (λ = 0.41266(2) Å) at the MSPD beam line, ALBA.31 Si and LaB6 were used as standard samples for calibration. Glass capillaries (∅ 0.5 mm) were used for the ex situ SRD test. In operando XAS measurements were performed at P65 of PETRA III in DESY. In operando XAS spectra were recorded in transmission geometry with the conventional step-scan mode at the Co K-edge during electrochemical cycling. The double-crystal fixed exit monochromator was equipped with the Si (111) crystal, and the ionization chambers were optimized for the Co K-edge. A multichannel potentiostat (VMP3, Bio-Logic) was supplied by beamline P02.1 (PETRA III, DESY) for electrochemical cycling. The spectra were processed by using the Demeter software.32
Results and discussion
Hydrothermal synthesis and characterization of the pure ZCO and ZCO/CC composite
The XRD pattern in Fig. 2a confirms that the ZCO precursor is composed of metal carbonates (CoCO3 and ZnCO3). During the hydrothermal process, Co2+ and Zn2+ ions can co-precipitate with carbonate in a weak alkaline environment (pH value of 6.0–9.0). Herein, urea can react with H2O and then converts to NH3 (NH4+) and CO2 (CO32−), which can provide the alkaline environment and serve as the precipitant, respectively. The overall reactions during the hydrothermal process can be, therefore, described as follows:
| (H2N)CO(NH2) + H2O → CO2 + 2NH3 (70 °C) |
| CO2 + 2NH3·H2O → CO32− + 2NH4+ |
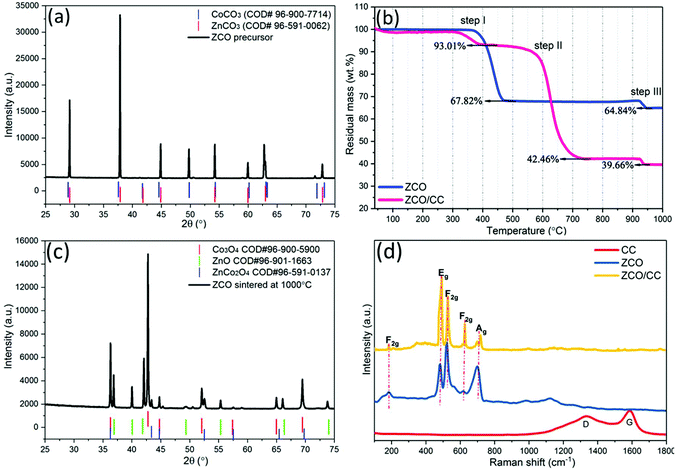 |
| | Fig. 2 XRD patterns (Co Kα1) of the ZCO precursor (a) before and (c) after the TG test; (b) TG curves of the ZCO precursor and the ZCO/CC precursor under oxygen/argon flow; (d) Raman spectra of the CC, ZCO and ZCO/CC. | |
To determine the optimal sintering temperature, TG analysis was performed from 35 °C to 1000 °C under oxygen/argon (∼1:3 in v:v) flow. In Fig. 2b, the ZCO precursor exhibits a major weight loss at 360–470 °C due to decomposition of the metal carbonates (CoCO3 at 350 °C and ZnCO3 at 400 °C). At 470 °C, 67.82 wt% of mass remains, which is in agreement with the theoretical value of 68.06 wt% (ZnCO3 + 2CoCO3 + 0.5O2 → ZnCo2O4 + 3CO2↑). Due to the presence of the CC, the mass loss of the ZCO/CC in step I is only 6.99 wt%, which is related to the decomposition of the Zn and Co-containing compounds. The ZCO/CC displays an additional step at 600–700 °C (step II) where the mass loss is 50.55 wt%. This is related to the combustion of the CC. Finally, the residual mass of 42.46 wt% at 750 °C can be associated with the amount of ZCO. Fig. 2c shows the presence of residual materials after step III (above 930 °C). This material is a mixture of ZnCo2O4, ZnO, Co3O4, and possibly CoAlO3 or ZnAl2O4, which is due to the decomposition of the ZCO (3ZnCo2O4 → 3ZnO + 2Co3O4 + 0.5O2↑) and contamination from the Al2O3 crucible, respectively. Based on these results, the sintering temperature was set at 500 °C, and the ZCO accounts for 45.65 wt% in the ZCO/CC composite after calcination treatment. The Raman spectrum of the CC (Fig. 2d) exhibits two distinct peaks located at 1336 cm−1 (D-band) and 1589 cm−1 (G-band), ascribed to the defective- and graphitized-carbon structure, respectively. However, these bands disappear in the spectrum of ZCO/CC, indicating the homogeneous surface covering of ZCO on the CC. Similar to the ZCO, the ZCO/CC shows also the characteristic bands located at 182, 487, 527, 625 and 717 cm−1, corresponding to the F2g, Eg, F2g, and A1g modes of ZCO, respectively.33,34 Based on the above experimental evidence, we can conclude that the ZCO powder and the ZCO/CC composite of homogeneous ZCO covered CC were successfully simultaneously synthesized via a hydrothermal reaction.
As shown in Fig. 3a, the pink rhombohedral ZCO precursor displays a wide size distribution (5–100 μm). However, the well-crystallized micrometer scale particles display a prismatic shape with a smooth exterior and distinct edges, apparently suggesting high crystallinity for these crystals. The sintering process can induce the removal of CO32− and volume change, leading to the pulverization of large particles into small ones (see Fig. 3a vs. b). The SEM image (Fig. 3c) of the ZCO particles shows that the material has a parallelepiped-like shape, with angles of α = 90°, β = 90° and γ = 150°. The formation mechanism of ZnCo2O4 with a similar morphology was also reported by Cheng et al.35 The particle shape is related to the hydrothermal conditions and the inorganic additive. As demonstrated by EDX mapping (see Fig. 3d–f and Fig. S1†), a fairly uniform elemental distribution of Zn, Co and O elements is observed over the entire particles. A long-time hydrothermal reaction can enable sufficient dissolution–precipitation reaction between metal ions and metal carbonate, forming phase-pure parallelepiped-like shaped ZCO particles.
 |
| | Fig. 3 SEM morphological characterization: (a) the ZCO precursor after the hydrothermal reaction; (b, c) the ZCO after being sintered at 450 °C; and EDX mapping of the ZCO: Zn (d), Co (e) and O (f). | |
The XRD pattern (MoKα1) of the as-obtained ZCO powders is shown in Fig. 4a. The Rietveld refinement confirms the main cubic phase (space group: Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, COD #96-591-0137, 94.90 wt%) and tiny hexagonal phase (space group: P63mc, COD #90-153-3021, 5.10 wt%) in the ZCO. Satisfactory agreement factors (Rwp = 8.55%, Rp = 6.32%) with the cell parameter a = 8.108 Å and cell volume = 533.052(6) Å3 were obtained in this crystal refinement. The cubic spinel ZnCo2O4 consists of the octahedral CoO6 and tetrahedral ZnO4 structure.36 As depicted in Fig. 4b, the reflections of the ZCO/CC are well indexed to the Fdm cubic phase ZCO (96.03 wt%), together with tiny P63mc hexagonal phase ZCO (3.97 wt%). Two broad and tiny diffraction peaks at ∼11° and ∼20° are due to the presence of the CC in the composite (see Fig. S2†). The surface chemical composition and chemical states of the ZCO were analyzed by XPS. The XPS spectra in Fig. 4c–f confirm the presence of Zn, Co and O. The Zn LMM Auger line at ∼989 eV (Fig. 4c) indicates the presence of Zn2+. Furthermore, two components can be observed in the Zn 2p3/2 spectrum (Fig. 4d), one at 1021.2 eV and one at 1022.7 eV, indicating the presence of Zn2+ in tetrahedral and octahedral oxygen coordination, respectively. The tiny amount of Zn2+ in the octahedral oxygen coordination is due to Zn2+ and Co3+ exchanging their positions.37 According to the previous work by Azmi et al.,38,39 the 2p spectrum of cobalt (Fig. 4e) was fit by considering the typically occurring multiplet splitting observed for transition metals. Similar to the results from Kumar et al.,40 the fit multiplet defined as in the study of Biesinger et al.,41 accompanied by the absence of the Co3+ satellite (785.6 eV), proves the presence of Co3+ only. The O 1s spectrum (Fig. 4f) can be resolved into three components, and the most intensive one is attributed to the oxygen in the metal oxide (529.9 eV), whereas C
m, COD #96-591-0137, 94.90 wt%) and tiny hexagonal phase (space group: P63mc, COD #90-153-3021, 5.10 wt%) in the ZCO. Satisfactory agreement factors (Rwp = 8.55%, Rp = 6.32%) with the cell parameter a = 8.108 Å and cell volume = 533.052(6) Å3 were obtained in this crystal refinement. The cubic spinel ZnCo2O4 consists of the octahedral CoO6 and tetrahedral ZnO4 structure.36 As depicted in Fig. 4b, the reflections of the ZCO/CC are well indexed to the Fdm cubic phase ZCO (96.03 wt%), together with tiny P63mc hexagonal phase ZCO (3.97 wt%). Two broad and tiny diffraction peaks at ∼11° and ∼20° are due to the presence of the CC in the composite (see Fig. S2†). The surface chemical composition and chemical states of the ZCO were analyzed by XPS. The XPS spectra in Fig. 4c–f confirm the presence of Zn, Co and O. The Zn LMM Auger line at ∼989 eV (Fig. 4c) indicates the presence of Zn2+. Furthermore, two components can be observed in the Zn 2p3/2 spectrum (Fig. 4d), one at 1021.2 eV and one at 1022.7 eV, indicating the presence of Zn2+ in tetrahedral and octahedral oxygen coordination, respectively. The tiny amount of Zn2+ in the octahedral oxygen coordination is due to Zn2+ and Co3+ exchanging their positions.37 According to the previous work by Azmi et al.,38,39 the 2p spectrum of cobalt (Fig. 4e) was fit by considering the typically occurring multiplet splitting observed for transition metals. Similar to the results from Kumar et al.,40 the fit multiplet defined as in the study of Biesinger et al.,41 accompanied by the absence of the Co3+ satellite (785.6 eV), proves the presence of Co3+ only. The O 1s spectrum (Fig. 4f) can be resolved into three components, and the most intensive one is attributed to the oxygen in the metal oxide (529.9 eV), whereas C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (531.6 eV) and C–O (532.7 eV) stem from contamination. The quantitative analysis of the spectra leads to a Zn:Co:O ratio of 1:2.3:3.9, which well agrees with the expected stoichiometry of the ZnCo2O4.
O (531.6 eV) and C–O (532.7 eV) stem from contamination. The quantitative analysis of the spectra leads to a Zn:Co:O ratio of 1:2.3:3.9, which well agrees with the expected stoichiometry of the ZnCo2O4.
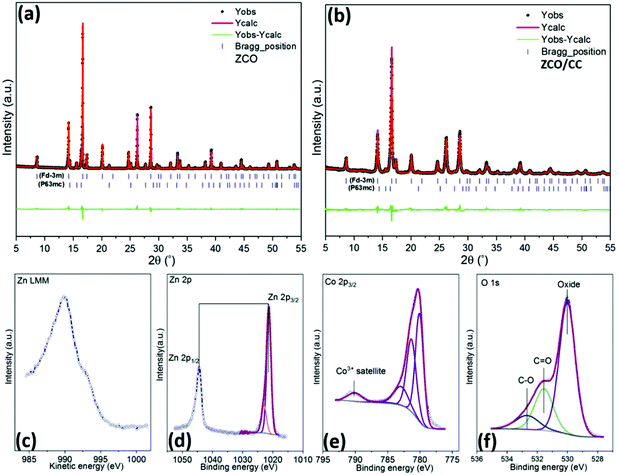 |
| | Fig. 4 The Rietveld refinement of the crystal structure of the ZCO powder (a) and ZCO/CC composite (b); and the XPS spectra of the pristine ZCO powder: (c) Zn LMM, (d) Zn 2p, (e) Co 2p3/2 and (f) O 1s. | |
Energy storage mechanism of the ZCO electrode
Fig. 5a shows the CV curves of the ZCO electrode over 50 cycles. Based on previous reports, during the first cathodic scan, the weak peak at 1.7 V is related to the initial Li+ insertion into the ZCO structure.42 Then, the CV shows an intensive irreversible peak at ∼0.7 V followed by a minor peak at ∼0.4 V, corresponding to the conversion of ZCO to metallic Zn and Co, and a further alloying of Zn with Li. In the following cycles, the main reduction peak (∼0.7 V) shifts to ∼1.0 V, a phenomenon which is in agreement with the literature.23 During the anodic potential sweep, Li dealloys from LiZn at ∼0.6 V, and the two peaks located at around 1.7 V and 2.2 V correspond to the formation of ZnO and CoO, respectively.5,43 After ten cycles, the cathodic peaks apparently shift to a lower potential, at ∼0.3 V, while the anodic peaks gradually disappear, indicating the decrease in reaction kinetics.44Fig. 5b shows the GC potential profiles recorded at 0.1 A g−1. The 1st lithiation and delithiation capacities are 1087 and 806 mA h g−1, respectively, which are beyond the theoretical values of 976 mA h g−1 and 789 mA h g−1 based on 9 and 7 Li+ pfu, respectively. The high irreversible capacity during the 1st cycle can be ascribed to the initial formation of the solid-electrolyte interphase (SEI) layer, as already observed on other conversion-type materials.29,45–47 A severe capacity fade is observed after 20 cycles, which can be mainly ascribed to the pulverization of the active material and reduction of electronic conductivity.
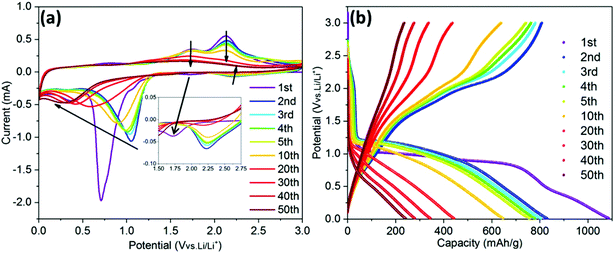 |
| | Fig. 5 The CV curves at a scan rate of 0.1 mV s−1 (a) and GC potential profiles recorded at 0.1 A g−1 (b) of the ZCO electrode. | |
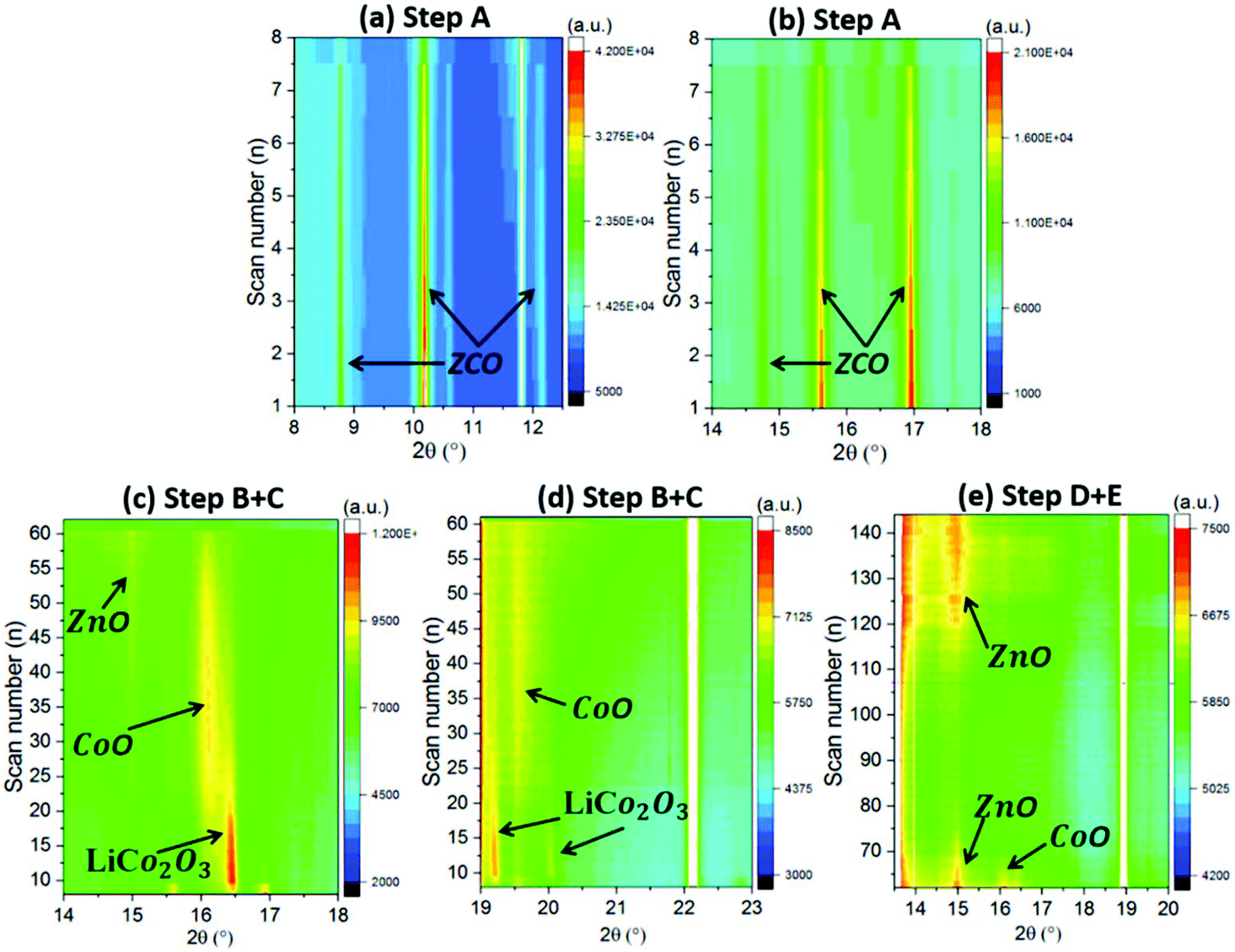
In order to further understand the electrochemical reaction mechanism of the ZCO, in operando SRD was performed during the 1st GC cycle at 75 mA g−1. Based on the above discussed electrochemical behavior (Fig. 5 and 6a), the appearance of potential plateaus in the GC and current peaks in the CVs can be related to specific phase changes. The SRD patterns, containing a total of 145 scans (see Fig. 6b), can be separated into five steps (named from A to E in Fig. 6 and 7). In step A (scan 1–7, Fig. 7a and b), where the potential rapidly drops and forms a short plateau at ∼1.16 V, the intensity of the ZCO peaks decreases slightly. In this region, the Li-ion intercalation into cubic ZCO leads to an increase of cell parameters (e.g., lattice parameter a: 8.108 Å at scan 1 → 8.111 Å at scan 3 → 8.113 Å at scan 5 → 8.114 Å at scan 7, see Fig. S3a†).
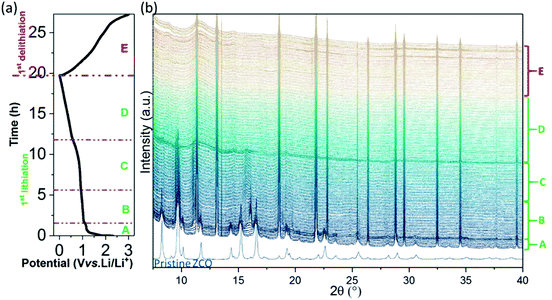 |
| | Fig. 6
In operando SRD measurements of the ZCO electrode: (a) potential profile and (b) SRD patterns during the 1st lithiation/delithiation. | |
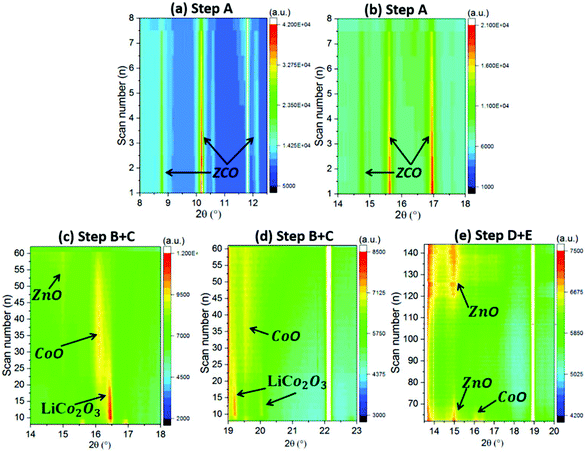 |
| | Fig. 7
In operando SRD contour map of the ZCO with different reaction steps: (a, b) step A, (c, d) step B + C and (e) step D + E. | |
Between step A and step B (scan 7–9), when there is a small steep potential slope (1.16 V → 1.07 V), a new series of XRD reflections appear at 9.8°, 11.3°, 16.1°, 18.8° and 19.7°, as depicted in Fig. 6a and Fig. S3c and d.† These peaks correspond to LiCo2O3 (Li0.33Co0.67O, COD #96-154-1452), indicating that the Li+ intercalated ZnCo2O4 (LiZnCo2O4) converts to LiCo2O3 and ZnO. Step B covers the scan 8–31, corresponding to the potential plateau at 1.05–0.93 V. As shown in Fig. 7c and d and the enclosed red dashed line of Fig. S3e and f,† the peak intensity of the LiCo2O3 reaches the maximum value at scan 10 and then gradually decreases until scan 31. Meanwhile, the intensity of the CoO reflections (at 9.6°, 11.1°, 15.7° and 19.3° enclosed in pink dashed line in Fig. S3e and f,† COD #96-154-1663) increases, indicating the conversion from LiCo2O3 to CoO and Li2O during Li+ insertion. Moreover, a series of weak reflections related to ZnO (at 8.4°, 9.1°, 14.6° and 17.3° in Fig. S3e and f,† COD #96-230-0114) can also be observed. Simultaneously, the diffraction peaks related to ZCO gradually weaken until they completely vanish at scan 17 at around 1.01 V. In step C (scan 32–63), corresponding to a potential plateau at 0.93–0.85 V (scan 48) and a gentle slope (0.85–0.59 V), the intensity of the new reflections (LiCo2O3, CoO and ZnO) decreases and finally vanishes (see Fig. 7c and d and Fig. S3g and h†). It relates to the continuous reduction reaction of CoO and ZnO to metallic Co and Zn. In step D (scan 64–106), related to a potential slope at 0.57–0.01 V, except for further vanishing of diffraction peaks related to residual LiCo2O3, CoO and ZnO, broad and weak peaks ascribed to Li2O and metallic Zn and Co are observed (see Fig. 7e and Fig. S4a and b†). The typical alloying reaction of Zn with Li occurs below 0.5 V, and the SEI forms at ∼0.8 V.28,48,49 Unfortunately, due to the nature of the nanoparticles produced by the electrochemical conversion reaction, the LiZn alloy cannot be observed in the XRD patterns. Herein, this is the first time that the multiple conversion processes are observed on a ZCO electrode. In general, the first conversion reaction is LiZnCo2O4 → LiCo2O3 + ZnO, followed by the second conversion of LiCo2O3 → CoO + Li2O, and finally, the CoO and ZnO are reduced to metallic Co and Zn (LiZn alloy) nanograins.
During the delithiation process (step E, scan 107–145), the dealloying reaction reversibly takes place at ∼0.6 V.28 At a potential above 1.34 V (scan 121), the XRD reflections located at 8.6°, 14.8° and 9.6° slightly increase in intensity (see Fig. 7e and Fig. S4c and d†), indicating the formation of the oxidation products, ZnO and CoO. The nanosized products block further analysis based on phase change during the continuous (de)lithiation process.
To clarify the evolution of the cobalt valence in the ZCO electrode, in operando XAS measurements were carried out at the Co K-edge during the 1st lithiation performed at 80 mA g−1 (see Fig. 8a). Fig. 8b depicts that Co3+ (scan 1) in the pristine electrode is gradually reduced to Co2+ (scan 6), corresponding to the step A + B as defined according to the in operando SRD test. During scan 1–2 (step A), the cobalt valence changes dramatically from higher than Co3O4 to lower than Co3O4, which is in agreement with the conversion from ZnCo2O4 to LiCo2O3. Later on, LiCo2O3 converts to CoO gradually during scan 3–6 (step B). From scan 7 to scan 11, CoO converts to Co continuously, which can be related to step C in the in operando SRD (see Fig. 8c). Finally, from scan 12 to scan 17 (see Fig. 8d), the spectra are overlapped and consistent with the spectrum of Co foil, indicating that the reduction from CoO to Co completes in step C. Therefore, the evolution of the cobalt valence is well in agreement with the in operando SRD results. Based on the combination of the in operando SRD and XAS analyses, the lithiation/delithiation process in the ZCO electrode is described in the following equations:
Step A (OCV ∼1.16 V):
| ZnCo2O4 + Li+ + e− → LiZnCo2O4 (∼1.16 V) |
Step B (1.16 ∼0.93 V):
| LiZnCo2O4 → LiCo2O3 + ZnO (1.16 ∼1.05 V) |
| LiCo2O3 + Li+ + e− → 2CoO + LiO2 (1.05 ∼0.93 V) |
Step C (0.93 ∼0.59 V):
| CoO + 2Li+ + 2e− → Co + Li2O |
| ZnO + 2Li+ + 2e− → Zn + Li2O |
Step D (0.57 ∼0.01 V):
Step E (0.01 ∼3.00 V):
| Zn + Li2O → ZnO + 2Li+ + 2e− |
| Co + Li2O → CoO + 2Li+ + 2e− |
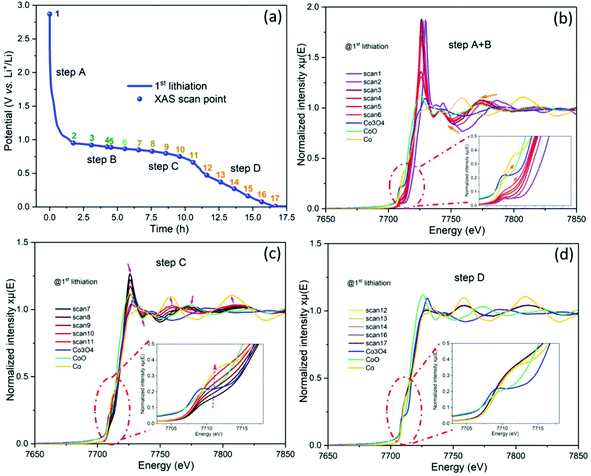 |
| | Fig. 8 (a) The potential profile of the ZCO half-cell during the in operando XAS with a current density of 80 mA g−1, and the NEXAS spectra on the Co K-edge during the 1st lithiation process: (b) step A + B, (c) step C and (d) step D. | |
ZCO/CC composite based flexible binder-free electrode
The flexible self-standing ZCO/CC composite electrode was prepared by a hydrothermal reaction. The XRD (Fig. 4b) and SEM (Fig. 9a) confirm the homogeneous growth of the crystal ZCO on the CC after the annealing of the ZCO/CC precursor. From the SEM image of Fig. 9b, one can observe the ZCO nanoflakes (∼116 nm thickness) grown on the fibers. The open space between the nanoflakes allows efficient accommodation of large volume changes and releases structural stress. From the inserted image in Fig. 9a, it is clear that the highly flexible and self-supported ZCO/CC electrode can suffer a 180° mechanical bending. In short, the hierarchical ZCO/CC composite as a self-supporting electrode can combine the advantages of high electronic conductivity, high surface-to-volume ratio and short Li-ion diffusion length.
 |
| | Fig. 9 SEM images of the ZCO/CC self-standing electrode: (a) a global map of the coated CC (the inset shows a bent electrode) and (b) a magnified image displaying ZCO nanoflakes. | |
To verify the improvement of the electrochemical performance of the ZCO nanoflakes grown on the 3D conductive substrate, CV measurements were carried out for both ZCO and ZCO/CC samples. Fig. 10a and b show that the main cathodic peaks of the ZCO are located at 1.0–0.5 V, corresponding to the reductions of Co2+ to Co0 and Zn2+ to Zn0. In addition, two anodic peaks at ∼1.8 V and ∼2.1 V appear during the delithiation process, ascribed to the oxidation reactions of Zn → ZnO and Co → CoO, respectively.50,51 In contrast, the ZCO/CC electrode exhibits additional redox reactions at a low potential (∼0.4 V at lithiation process/∼0.6 V at delithiation process), which can be attributed to Li+ storage in the CC. Moreover, the anodic/cathodic peaks shift towards each other slightly in the ZCO/CC electrode compared with the ZCO one, indicating a lower polarization induced by the enhanced electronic conductivity. Different from the opinion of Liu et al.,25 the contribution of the CC to the capacity (∼80 mA h g−1 at 0.1 A g−1, see Fig. S5†) is not negligible.
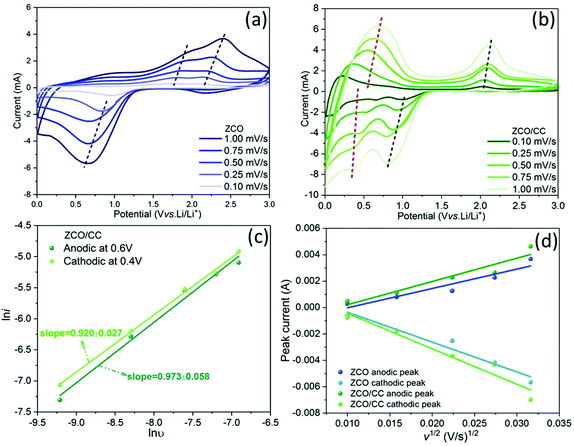 |
| | Fig. 10 CV curves of the ZCO (a) and the ZCO/CC (b) at different scan rates of 0.1–1.0 mV s−1; (c) power-law dependence of generated currents on scan rates and corresponding linear fitting of the pair of redox peaks at ∼0.4 V (cathodic) and ∼0.6 V (anodic) for the ZCO/CC; (d) profiles of the peak current (ip) vs. the square root of the scan rate (ν1/2) and the corresponding linear fitting for the ZCO and the ZCO/CC. | |
Herein, two mechanisms are considered to elucidate the Li+ storage in the CC: (i) Li+ intercalation into graphitized carbon to form LiC6 and (ii) Li+ storage on the carbon surface via the capacitive effect.52,53 The two effects can be distinguished by analyzing the power-law dependence relationship between the generated current (i) and scan rate (ν) from the CV data (iV = aνb).54Fig. 10c shows the linear fitting according to the relationship of lniV = lna + blnv. The fitting result for both the cathodic peak at ∼0.4 V (b = 0.920 ± 0.027) and the anodic peak at ∼0.6 V (b = 0.973 ± 0.058) for the ZCO/CC displays a b value of around 1.0, meaning that the contribution of the capacitive charge storage in the CC is the main reason for the enhanced capacity at low potential. Fig. 10a and b show that voltage hysteresis of the CV peaks is gradually enlarged as the scan rate increases. To further investigate the reaction kinetics, the Li+ diffusion coefficient is calculated from the peak currents based on the Randles–Sevcik equation:55
| | | ip2 = 0.2n3F3c2A2DLiν/(RT) | (1) |
where
ip is the peak current (A),
n is the number of electrons transferred in the redox reaction,
F is Faraday constant (96
485 C mol
−1),
c is the concentration of Li
+ species (0.15 mol cm
−3 for the ZCO),
A is the effective contact area between ZCO and the electrolyte,
DLi is the Li
+ diffusion coefficient (cm
2 s
−1),
ν is the scan rate (V s
−1),
R is the gas constant (8.314 J mol
−1 K
−1) and
T is the absolute temperature (298 K). As shown in
Fig. 10d, the linear relationship between peak currents (
ip) and the square root of the scan rate (
ν1/2) suggests that the electrochemical storage process is limited by the diffusion process. Because the effective contact area (
A) of these compounds is rather complex due to the huge difference in morphology between the ZCO particles and the ZCO/CC composite, we propose to use a comprehensive parameter (
AD1/2Li (cm
3 s
−1/2)) to describe the apparent effective diffusion in the solid phase. From this calculation,
AD1/2Li related to the ZCO/CC is 1.2 times higher than the one related to the pure ZCO (anodic: 2.48 × 10
−7vs. 2.97 × 10
−7 cm
3 s
−1/2; cathodic: 3.81 × 10
−7vs. 4.53 × 10
−7 cm
3 s
−1/2 for the ZCO
vs. ZCO/CC, see Table S1
†).
Fig. 11a compares the rate capability of the ZCO and the ZCO/CC at different current densities from 0.1 to 1.0 A g−1, whereas the capacity decreases with rising current density due to the sluggish Li+ diffusion kinetics.56 For a fair comparison, the capacity contribution from the CC in the ZCO/CC electrodes is not included, by subtracting the capacity of pure CC (see Fig. S5†). The ZCO/CC electrode exhibits a much better rate performance (delithiation capacity: 1013, 948, 760, 664 and 603 mA h g−1 at 0.1, 0.25, 0.5, 0.75 and 1.0 A g−1, respectively) compared to the ZCO electrode, while the ZCO displays capacities of 1049, 832, 451, 249 and 131 mA h g−1 at 0.1, 0.25, 0.5, 0.75 and 1.0 A g−1, respectively. When the current density is reduced again to 0.1 A g−1, the capacity of the ZCO/CC electrode (1207 mA h g−1) is beyond the original value. Furthermore, in commercial application, the specific capacity of cells should be valued considering the total mass of all cell parts. Actually, the mass of inactive components in the electrode, such as binder, conductive additive and current collector, should be also taken into account. In this work, the apparent specific capacities of the ZCO and the ZCO/CC electrodes are also compared based on the total electrode mass, namely ZCO + carbon black + polymer binder + copper foil and ZCO + CC, respectively. As shown in Fig. S6,† the apparent specific capacity of the ZCO/CC is 296 mA h g−1 at 0.1 mA g−1, which is almost twice the value of the ZCO (170 mA h g−1). At higher current densities, the disparity becomes even wider (176 mA h g−1 for the ZCO/CC vs. 22 mA h g−1 for the ZCO at a current density of 1.0 A g−1). This is not only due to the light weight of the CC but also to its contribution to providing extra energy storage at low potential and enhancing electronic conductivity. This result indicates the potential of such a carbon cloth supported flexible electrode on improving the electrochemical performance and enhancing the apparent specific capacity.
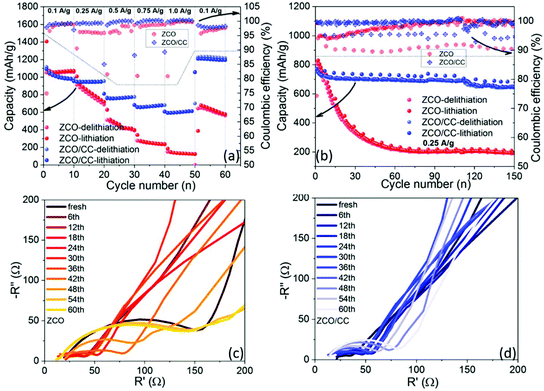 |
| | Fig. 11 Electrochemical performance of the ZCO and ZCO/CC: the rate capability (a), the cycling stability at 0.25 A g−1 (b), and the selected Nyquist plots of the ZCO (c) and ZCO/CC (d) during the GC test at 3 V. | |
The long-term GC test performed with a current density of 0.25 A g−1 is presented in Fig. 11b. The ZCO displays serious capacity loss within 60 cycles, while the capacity of the ZCO/CC is still stable until the ∼110th cycle. The delithiation capacities at the 60th cycle of the ZCO/CC and ZCO electrodes are around 701 and 225 mA h g−1, respectively. It is also noteworthy that the coulombic efficiency of the ZCO/CC is always higher than that of the ZCO in both the rate test and cycling test. Therefore, it is confirmed by the improved reaction reversibility the benefits of the combination of the conductive matrix and ZCO nanoflakes, which not only allows the progressive penetration of the liquid electrolyte into the interior ZCO but also facilitates electron conduction through the CC. To examine the resistance evolution during the long-term cycling test, the EIS plots were recorded every six cycles at 3.0 V during the cycling test. As shown in Fig. 11c and d, a Nyquist plot consists of a semicircle at medium frequency and an inclined line at low frequency, related to the interface resistance and Li-ion diffusion in the electrode, respectively. The ZCO electrode displays a high interface resistance in the pristine state, which apparently decreases after the 1st cycle due to the formation of the ionic conductive SEI layer and the larger active surface area induced by the initial conversion. The resistance dramatically increases after 36 cycles, due to the much thicker SEI layer and excessive electrode pulverization.57 In comparison, the interface resistance of the ZCO/CC electrode is low at the beginning and only increases slightly during long-term cycling, suggesting a fast reaction kinetics, which can be related to the well-maintained integrity of active particles and fast Li+ and electron diffusion in the ZCO/CC electrode.28,47–49,58 Such attractive properties are ascribed to the unique morphology of this binder-free electrode.
Conclusions
In this study, phase pure ZnCo2O4 was prepared by a hydrothermal method and used as an anode material for LIBs. The mechanism of Li storage in spinel ZCO is investigated by in operando SRD and XAS during galvanostatic cycling. Electrochemical intercalation, conversion and alloying reactions of the ZCO electrode are discussed in detail. It is the first time that the intermediate phases (LiCo2O3 (at 1.16 V), CoO (at ∼0.9 V) and ZnO (at ∼1.1 V)) are evidenced during the 1st lithiation process. This is similar to our study on the ZnMn2O4 anode, where LiZnMn2O4 is formed during the initial lithium storage. After the formation of CoO and ZnO, the two oxides can be further converted to metallic Zn (or LiZn), Co and Li2O. In contrast to the previous conclusion, during the first delithiation, CoO instead of Co3O4 is formed. As a binder-free anode for LIBs, the hierarchical ZCO/CC delivers high specific capacity and shows excellent cycling stability (701 mA h g−1 at the 60th cycle at a specific current of 0.25 A g−1). This appealing electrochemical behavior can be attributed to the large electrochemical active area and fast ion/electron transfer in the hierarchical ZCO/CC composite.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Authors acknowledge the financial support of this research by the China Scholarship Council. Authors thank prof. Helmut Ehrenberg for his support on this work and for the fruitful discussions. Authors also appreciate the technical support from colleagues of IAM-ESS, KIT: Qiang Fu and Angelina Sarapulova for the in operando synchrotron SRD and XAS tests, Liuda Mereacre for the Raman test, Bettina Hunzinger for the SEM measurement. The multichannel potentiostat (VMP3, Bio-Logic) for in operando XAS measurement was supplied by beamline P02.1 PETRA III, DESY. The K-Alpha+instrument was supplied by the Federal Ministry of Economics and Technology on the basis of a decision by the German Bundestag. This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe).
References
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- M. Peng, B. Dong and D. Zou, J. Energy Chem., 2018, 27, 611–621 CrossRef.
- J. Bai, K. Wang, J. Feng and S. Xiong, ACS Appl. Mater. Interfaces, 2015, 7, 22848–22857 CrossRef CAS PubMed.
- F. Cheng, Z. Tao, J. Liang and J. Chen, Chem. Mater., 2008, 20, 667–681 CrossRef CAS.
- Y. Sharma, N. Sharma, G. V. Subba Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2855–2861 CrossRef CAS.
- J. Wang, H. Wang, F. Li, S. Xie, G. Xu, Y. She, M. K. H. Leung and T. Liu, J. Mater. Chem. A, 2019, 7, 3024–3030 RSC.
- Q. Q. Ren, F. Da Yu, S. W. Zhang, B. S. Yin, Z. B. Wang and K. Ke, Electrochim. Acta, 2019, 297, 1011–1017 CrossRef CAS.
- K. Liu, F. Zou, Y. Sun, Z. Yu, X. Liu, L. Zhou, Y. Xia, B. D. Vogt and Y. Zhu, J. Power Sources, 2018, 395, 92–97 CrossRef CAS.
- J. Lu, Z. Chen, F. Pan, Y. Cui and K. Amine, Electrochem. Energy Rev., 2018, 1, 35–53 CrossRef.
- Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- Y. Lu, L. Yu and X. W. (David) Lou, Chem, 2018, 4, 972–996 CAS.
- M. R. Gao, Y. F. Xu, J. Jiang and S. H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- X. Bin Zhong, H. Y. Wang, Z. Z. Yang, B. Jin and Q. C. Jiang, J. Power Sources, 2015, 296, 298–304 CrossRef.
- L. Hu, B. Qu, C. Li, Y. Chen, L. Mei, D. Lei, L. Chen, Q. Li and T. Wang, J. Mater. Chem. A, 2013, 1, 5596–5602 RSC.
- J. Deng, X. Yu, X. Qin, D. Zhou, L. Zhang, H. Duan, F. Kang, B. Li and G. Wang, Adv. Energy Mater., 2019, 1803612, 1803612 CrossRef.
- G. Balachandran, D. Dixon, N. Bramnik, A. Bhaskar, M. Yavuz, L. Pfaffmann, F. Scheiba, S. Mangold and H. Ehrenberg, ChemElectroChem, 2015, 2, 1510–1518 CrossRef CAS.
- K. Wu, D. Liu and Y. Tang, Electrochim. Acta, 2018, 263, 515–523 CrossRef CAS.
- L. Zhang, T. Wei, Z. Jiang, C. Liu, H. Jiang, J. Chang, L. Sheng, Q. Zhou, L. Yuan and Z. Fan, Nano Energy, 2018, 48, 238–247 CrossRef CAS.
- G. Zhang and X. W. (David) Lou, Sci. Rep., 2013, 3, 2–7 Search PubMed.
- Y. Luo, R. Guo, T. Li, F. Li, L. Meng, Z. Yang, Y. Wan and H. Luo, ChemElectroChem, 2019, 6, 690–699 CrossRef CAS.
- Y. Wang, P. Liu, K. Zhu, J. Wang, K. Yan and J. Liu, Electrochim. Acta, 2018, 273, 1–9 CrossRef CAS.
- B. Liu, J. Zhang, X. Wang, G. Chen, D. Chen, C. Zhou and G. Shen, Nano Lett., 2012, 12, 3005–3011 CrossRef CAS PubMed.
- L. Huang, G. H. Waller, Y. Ding, D. Chen, D. Ding, P. Xi, Z. L. Wang and M. Liu, Nano Energy, 2015, 11, 64–70 CrossRef CAS.
- Z. Wang, Q. Ru, Y. Mo, L. Guo, X. Chen, X. Hou and S. Hu, J. Mater. Sci.: Mater. Electron., 2017, 28, 9081–9090 CrossRef CAS.
- B. Liu, X. Wang, B. Liu, Q. Wang, D. Tan, W. Song, X. Hou, D. Chen and G. Shen, Nano Res., 2013, 6, 525–534 CrossRef CAS.
- J. Zhang, R. Chu, Y. Chen, H. Jiang, Y. Zhang, N. M. Huang and H. Guo, Ceram. Int., 2018, 44, 16219–16226 CrossRef CAS.
- W. Bai, H. Tong, Z. Gao, S. Yue, S. Xing, S. Dong, L. Shen, J. He, X. Zhang and Y. Liang, J. Mater. Chem. A, 2015, 3, 21891–21898 RSC.
- Y. Ma, Y. Ma, D. Geiger, U. Kaiser, H. Zhang, G. T. Kim, T. Diemant, R. J. Behm, A. Varzi and S. Passerini, Nano Energy, 2017, 42, 341–352 CrossRef CAS.
- Z. Zhao, G. Tian, A. Sarapulova, V. Trouillet, Q. Fu, U. Geckle, H. Ehrenberg and S. Dsoke, J. Mater. Chem. A, 2018, 6, 19381–19392 RSC.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- M. Herklotz, J. Weiß, E. Ahrens, M. Yavuz, L. Mereacre, N. Kiziltas-Yavuz, C. Dräger, H. Ehrenberg, J. Eckert, F. Fauth, L. Giebeler and M. Knapp, J. Appl. Crystallogr., 2016, 49, 340–345 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- T. Yu, Y. W. Zhu, X. J. Xu, Z. X. Shen, P. Chen, C.-T. Lim, J. T.-L. Thong and C.-H. Sow, Adv. Mater., 2005, 17, 1595–1599 CrossRef CAS.
- S. Cheng, H. Rong, Z. Jiang, Z. Zhen, G. Xie, B. Chen, B. Deng, J. Huang and Z.-J. Jiang, Ionics, 2017, 23, 77–85 CrossRef CAS.
- S. Cheng, H. Rong, Z. Jiang, Z. Zhen, G. Xie, B. Chen, B. Deng, J. Huang and Z.-J. Jiang, Ionics, 2017, 23, 77–85 CrossRef CAS.
- J. Bai, X. Li, G. Liu, Y. Qian and S. Xiong, Adv. Funct. Mater., 2014, 24, 3012–3020 CrossRef CAS.
- V. Šepelák, S. M. Becker, I. Bergmann, S. Indris, M. Scheuermann, A. Feldhoff, C. Kübel, M. Bruns, N. Stürzl, A. S. Ulrich, M. Ghafari, H. Hahn, C. P. Grey, K. D. Becker and P. Heitjans, J. Mater. Chem., 2012, 22, 3117 RSC.
- R. Azmi, V. Trouillet, M. Strafela, S. Ulrich, H. Ehrenberg and M. Bruns, Surf. Interface Anal., 2018, 50, 43–51 CrossRef CAS.
- R. Azmi, M. Masoumi, H. Ehrenberg, V. Trouillet and M. Bruns, Surf. Interface Anal., 2018, 1–6 Search PubMed.
- V. Kumar, C. R. Mariappan, R. Azmi, D. Moock, S. Indris, M. Bruns, H. Ehrenberg and G. V. Prakashe, ACS Omega, 2017, 2, 6003–6013 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- X.-B. Zhong, H.-Y. Wang, Z.-Z. Yang, B. Jin and Q.-C. Jiang, J. Power Sources, 2015, 296, 298–304 CrossRef CAS.
- C. Ai, M. Yin, C. Wang and J. Sun, J. Mater. Sci., 2004, 39, 1077–1079 CrossRef CAS.
- S. W. Lee, C. W. Lee, S. B. Yoon, M. S. Kim, J. H. Jeong, K. W. Nam, K. C. Roh and K. B. Kim, J. Power Sources, 2016, 312, 207–215 CrossRef CAS.
- J.-M. Tarascon, P. Poizot, S. Laruelle, S. Grugeon and L. Dupont, Nature, 2000, 407, 496–499 CrossRef PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- H. Duncan, F. M. Courtel and Y. Abu-Lebdeh, J. Electrochem. Soc., 2015, 162, A7110–A7117 CrossRef CAS.
- M. S. Song, Y. J. Cho, D. Y. Yoon, S. Nahm, S. H. Oh, K. Woo, J. M. Ko and W. Il Cho, Electrochim. Acta, 2014, 137, 266–272 CrossRef CAS.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513–523 CrossRef CAS.
- N. Du, H. Zhang, B. D. Chen, J. B. Wu, X. Y. Ma, Z. H. Liu, Y. Q. Zhang, D. R. Yang, X. H. Huang and J. P. Tu, Adv. Mater., 2007, 19, 4505–4509 CrossRef CAS.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- P. Yu, C. Li and X. Guo, J. Phys. Chem. C, 2014, 118, 10616–10624 CrossRef CAS.
- A. P. Cohn, L. Oakes, R. Carter, S. Chatterjee, A. S. Westover, K. Share and C. L. Pint, Nanoscale, 2014, 6, 4669–4675 RSC.
- G. A. Muller, J. B. Cook, H. S. Kim, S. H. Tolbert and B. Dunn, Nano Lett., 2015, 15, 1911–1917 CrossRef CAS PubMed.
- D. Y. W. Yu, C. Fietzek, W. Weydanz, K. Donoue, T. Inoue, H. Kurokawa and S. Fujitani, J. Electrochem. Soc., 2007, 154, A253 CrossRef CAS.
- Q. Zhang, S.-Z. Huang, J. Jin, J. Liu, Y. Li, H.-E. Wang, L.-H. Chen, B.-J. Wang and B.-L. Su, Sci. Rep., 2016, 6, 25942 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, 170–192 CrossRef PubMed.
- C. Yuan, L. Zhang, L. Hou, L. Zhou, G. Pang and L. Lian, Chem. – Eur. J., 2015, 21, 1262–1268 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qi00356h |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Guiying
Tian
a,
Guiying
Tian