
Metal–organic frameworks based on polynuclear lanthanide complexes and octahedral rhenium clusters†
 *a
Jan
van Leusen,
b
Denis G.
Samsonenko,
ac
Vladimir A.
Lazarenko,
d
Yan V.
Zubavichus,
e
Paul
Kögerler
b
and
Yuri V.
Mironov
ac
*a
Jan
van Leusen,
b
Denis G.
Samsonenko,
ac
Vladimir A.
Lazarenko,
d
Yan V.
Zubavichus,
e
Paul
Kögerler
b
and
Yuri V.
Mironov
ac
Abstract
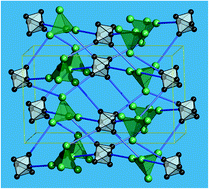
The octahedral chalcocyanide cluster anion [Re6S8(CN)6]4– can serve as a metallolinker for the self-assembly of metal–organic frameworks (MOFs) comprising polynuclear lanthanide complexes, which have been illustrated for a series of nine compounds isolated as [{Ln4(μ3-OH)4(ina)4(H2O)n}{Re6S8(CN)6}]·mH2O (Ln = Pr – Er, ina = isonicotinate). Their solid-state framework structures are based on {Ln4(μ3-OH)4}8+ cubane fragments interlinked by isonicotinate and rhenium cyanide clusters. The coordination environment of the lanthanide ions, the coordination numbers and even the number of bridging isonicotinate moieties changes systematically within the lanthanide row, however, the framework topology remains unchanged, confirming the structure-directing role of the rhenium cluster. Structures, magnetochemical properties, luminescence and thermal stability of the new MOFs have been thoroughly analyzed.
 Please wait while we load your content...
Please wait while we load your content...

