
Exploring wet chemistry approaches to ZnFe2O4 spinel ferrite nanoparticles with different inversion degrees: a comparative study†
Paolo
Dolcet,  *ab
Roland
Marschall,
ce
Stefano
Diodati,
a
Rafael
Muñoz-Espí,
f
Katharina
Landfester
d
and
Silvia
Gross
*a
*ab
Roland
Marschall,
ce
Stefano
Diodati,
a
Rafael
Muñoz-Espí,
f
Katharina
Landfester
d
and
Silvia
Gross
*a
*ab
Roland
Marschall,
ce
Stefano
Diodati,
a
Rafael
Muñoz-Espí,
f
Katharina
Landfester
d
and
Silvia
Gross
*a
Abstract
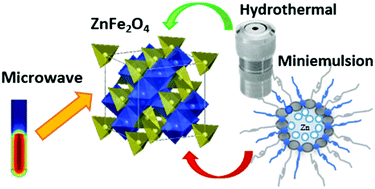
Zinc ferrite (ZnFe2O4) spinel nanocrystals were synthesised through three different low-temperature routes, namely hydrothermal, combined miniemulsion/hydrothermal, and microwave-assisted non-aqueous sol–gel synthesis. With the aim of studying the influence of the respective approach on the structural evolution of the compounds during the synthetic process, the samples were prepared with different processing times. The resulting nanoparticles were inspected with a wide range of analytical techniques, both with regards to the local (X-ray absorption spectroscopy – XAS) and the long-range (Raman spectroscopy, X-ray diffraction – XRD) order.
 Please wait while we load your content...
Please wait while we load your content...

