
Syntheses, crystal structures and magnetic properties of a series of luminescent lanthanide complexes containing neutral tetradentate phenanthroline-amide ligands†

Abstract
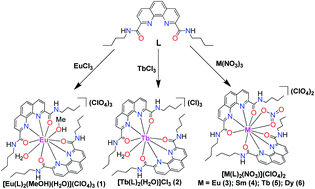
A series of lanthanide (Ln) compounds, [EuIII(L)2(MeOH)(H2O)](ClO4)3 (1), [TbIII(L)2(H2O)](Cl3) (2) and [MIII(L)2(NO3)](ClO4)2 (M = Eu (3); Sm (4); Tb (5); Dy (6)), have been prepared by the treatment of a neutral tetra-dentate ligand (N2,N9-dibutyl-1,10-phenanthroline-2,9-dicarboxamide, L) with various Ln(III) salts in MeOH. All of the compounds have been structurally characterized by X-ray crystallography. It is noted that the anions of the Ln(III) salts play crucial roles in the solid-state structures and the properties of the resulting complexes. In compounds 1 and 2, the metal centres are 10- and 9-coordinated, respectively, which are surrounded by two L ligands and solvent molecules. When the nitrate salts are used instead, four isostructural 10-coordinate Ln(III) compounds are obtained. The photophysical properties show that most of the compounds exhibit the characteristic emission peaks of Ln(III). Ligand L acts as a very good ‘antenna’ to sensitize the Eu(III) emission, but not for other Ln(III) emissions. The quantum yield of 3 is 75.4%, which is much higher than that of 1 (17.6%) due to the –OH oscillators of the coordinated H2O and MeOH molecules in 1. The magnetic properties of the Tb(III) compounds 2 and 5 and the Dy(III) compound 6 were investigated in detail as a natural extension of our previous study of the effect of high-coordination number on the single-ion magnet (SIM) behaviour of 3d compounds. The results show that subtle changes in the coordination environment have significant effects on the magnetic behaviour. The 9-coordinate compound 2 is not a single ion magnet (SIM), but both 10-coordinate compounds 5 and 6 exhibit field-induced SIM behaviour. It is also interesting to note that, upon the application of a small dc field, 5 exhibits two distinct relaxation processes, which is uncommon in Ln-based SIMs.
 Please wait while we load your content...
Please wait while we load your content...

