
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Field-induced slow relaxation of magnetization in the S = 3/2 octahedral complexes trans-[Co{(OPPh2)(EPPh2)N}2(dmf)2], E = S, Se: effects of Co–Se vs. Co–S coordination†
Eleftherios
Ferentinos‡
a,
Meixing
Xu‡
b,
Alexios
Grigoropoulos§
 a,
Ioannis
Bratsos
c,
Catherine P.
Raptopoulou
c,
Vassilis
Psycharis
c,
Shang-Da
Jiang
*bd and
Panayotis
Kyritsis
*a
a,
Ioannis
Bratsos
c,
Catherine P.
Raptopoulou
c,
Vassilis
Psycharis
c,
Shang-Da
Jiang
*bd and
Panayotis
Kyritsis
*a
aInorganic Chemistry Laboratory, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, GR-15771 Athens, Greece. E-mail: kyritsis@chem.uoa.gr
bCollege of Chemistry and Molecular Engineering, Beijing National Laboratory for Molecular Sciences, Beijing Key Laboratory of Magnetoelectric Materials and Devices, Peking University, Beijing 100871, P. R. China. E-mail: jiangsd@pku.edu.cn
cNCSR “Demokritos”, Institute of Nanoscience and Nanotechnology, 15310 Aghia Paraskevi, Athens, Greece
dBeijing Academy of Quantum Information Sciences, West Bld.#3, No. 10 Xibeiwang East Rd., Haidian District, Beijing 100193, P. R. China
First published on 12th April 2019
Abstract
The synthesis and the structural characterization of the octahedral trans-[Co{(OPPh2)(EPPh2)N}2(dmf)2], E = S (1), Se (2), complexes is described. These complexes are formed by crystallization of the corresponding tetrahedral [Co{(OPPh2)(EPPh2)N}2] complexes in the presence of dimethylformamide (dmf) and are the first examples of intact octahedral Co(II) complexes bearing dichalcogenidoimidodiphosphinato ligands. X-ray crystallography studies revealed a trans-CoO4E2 first coordination sphere consisting of two chelating (OPPh2)(EPPh2)N− ligands, as well as two dmf molecules coordinated via their O atoms. The isomorphous complexes 1 and 2 were studied by Direct Current magnetometry and shown to exhibit axial magnetic anisotropy. This finding was unequivocally confirmed by magnetic susceptibility measurements on an oriented crystal of complex 1, which clearly defined its easy axis of magnetization. Alternating Current magnetometry studies revealed field-induced slow relaxation of magnetization for both complexes, the characteristics of which are compared with those of other octahedral Co(II) complexes. The effects of the Se- vs. S-coordination on the magnetic anisotropy and relaxation properties of these and other 3d metal complexes are discussed.
Introduction
Ever since their discovery in the early 1990s,1,2 single-molecule magnets (SMMs) have attracted great interest,3 due to their potential applications in high-density data storage, molecular spintronics and quantum computation devices,4 as well as their interesting magnetothermal properties.5 These compounds exhibit slow relaxation of their magnetization and magnetic hysteresis below the corresponding blocking temperature, of pure molecular origin. The multinuclear [Mn12O12(CH3CO2)16(H2O)4] complex is the first molecular compound shown to exhibit an activation barrier (Ueff) for the relaxation of its magnetization.1,2,6 Following this observation, numerous multinuclear complexes, involving 3d metal ions, have been explored.7,8 The magnitude of Ueff in a SMM is controlled by the total spin, S, of the ground state, and the magnetic anisotropy of the system, expressed via the axial D zero-field splitting (zfs) component. Specifically, Ueff is tentatively considered to be equal to S2|D| or (S2 − 1/4)|D| for integer and non-integer spin systems, respectively. Therefore, the desired characteristics of SMMs are a large ground spin state S and a large magnetic anisotropy, D. Large S can be achieved via predominant ferromagnetic interactions in multinuclear metal complexes,9,10 but, due to their overall high symmetry, such systems exhibit remarkably small magnetic anisotropies (i.e. usually D < 1 cm−1).11 On the other hand, the principles of synthetic coordination chemistry, concerning the interplay between the metal ion and the ligands employed, can be applied much more effectively on mononuclear complexes, potentially leading to coordination spheres of the desired structural and, hence, magnetic properties.12 For that reason, research efforts have been recently directed towards the synthesis of mononuclear complexes of lanthanides13,14 and actinides,15 which exhibit remarkably large magnetic anisotropies, owing to large and unquenched spin–orbit coupling (SOC). Moreover, in some complexes of this type, blocking temperatures approaching that of liquid nitrogen (77 K) have been achieved16–18 thus opening up the prospects of potential technological applications.In an effort to master a synthetic control over the magnitude of D, a large number of 3d-based mononuclear complexes have been under extensive experimental19–22 or computational23,24 investigation. A trigonal pyramidal high spin Fe(II) complex is the first 3d metal-based mononuclear complex shown to exhibit field-induced slow relaxation of its magnetization.25 Following that report, slow relaxation of magnetization has been established for complexes of Cr(II), Mn(III), Mn(IV), Fe(I), Fe(II), Fe(III), Co(I), Co(II), Ni(I), Ni(II), Cu(II), Cu(III), Re(IV),19–22 and more recently of V(IV),26,27 Mn(II)28,29 and Ru(III).30
Complexes of Co(II) constitute the largest family of 3d-metal-based complexes showing slow relaxation of their magnetization.22,31 It should be stressed that the tetrahedral S = 3/2 (PPh4)2[Co(SPh)4] complex32,33 bearing a Co(II)S4 core, was the first mononuclear 3d metal complex to exhibit slow relaxation of its magnetization in the absence of a Direct Current (DC) magnetic field.34–37 Along these lines, tetrahedral [Co{(EPiPr2)2N}2], E = S,38 Se,38 Te,39 complexes, bearing dichalcogenidoimidodiphosphinato ligands40 and Co(II)E4 cores, exhibit slow relaxation of their magnetization, which was also observed in the absence of DC magnetic field for the E = Se, Te, complexes. On the other hand, high-spin, S = 3/2, octahedral Co(II) complexes exhibit magnetic anisotropy that can be tuned through appropriate structural modifications, for instance axial elongation or compression of an octahedral coordination sphere, leading to axial anisotropy (very small Δrh) and the presence of either easy axis (Δax < 0) or easy plane (Δax > 0), of magnetization, respectively (Δax is the axial and Δrh the rhombic crystal field parameter, vide infra).41 Ever since the [Co(dmphen)2(NCS)2] complex (dmphen = 2,9-dimethyl-1,10-phenanthroline) has been shown to exhibit field-induced slow relaxation of its magnetization,42 the relaxation properties of a large number of octahedral high-spin Co(II) complexes, exhibiting coordination spheres dominated Co–N or Co–O bonds, have been investigated (Table S1†).
Some of us have reported the conversion of the tetrahedral complexes [Ni{(OPPh2)(EPPh2)N}2], E = S,43 Se,44 to octahedral trans-[Ni{(OPPh2)(EPPh2)N}2(sol)2], sol = dmf,44 thf,45 dmso.46 Accurate zfs parameters for the latter S = 1 complexes were determined by high-frequency and -field EPR spectroscopy (HFEPR).45,46 In the work presented herein, the analogous conversion of tetrahedral [Co{(OPPh2)(EPPh2)N}2] E = S,47 Se,44 to octahedral trans-[Co{(OPPh2)(EPPh2)N}2(dmf)2] E = S (1), Se (2), is reported. Studies on the S = 3/2 complexes 1 and 2 by DC and Alternating Current (AC) magnetometry reveals that they exhibit field-induced slow relaxation of magnetization. The observed effects of the nature of the donor atoms E = S or Se, on the magnetic behavior of complexes 1 and 2, are discussed, and comparisons with other octahedral Co(II) complexes exhibiting field-induced slow relaxation of magnetization are made.
Results and discussion
Synthesis and IR spectroscopic characterization
Complexes 1 and 2 were obtained as crystalline products, upon slow diffusion of the coordinating solvent dmf into dichloromethane solutions of tetrahedral [Co{(OPPh2)(EPPh2)N}2], E = S,47 Se.44 The same procedure has been previously employed for the preparation of the analogous Ni(II) octahedral complexes trans-[Ni{(OPPh2)(EPPh2)N}2(dmf)2].44 The most characteristic IR bands of complexes 1 and 2 (Fig. S1 and S2,† respectively), as well as their comparison with those of the corresponding ligands, are shown and analyzed in the ESI.†X-ray crystallography
Single crystal X-ray diffraction revealed that complexes 1 and 2 are isomorphous (crystallographic data for both compounds are listed in Table S2†). A detailed crystallographic description is presented for complex 1 and, in parallel, the basic characteristics of complex 2 are also discussed. The molecular structure of 1 is shown in Fig. 1a and that of 2 in Fig. S3.† The isomorphous relationship between 1 and 2 is further supported by the overlay plot presented in Fig. 1b. Bond distance and bond angle values of 1 and 2 are listed in Table 1. Both compounds crystallize in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and possess a center of symmetry, occupied by a Co(II) ion. The latter is coordinated to two (OPPh2)(EPPh2)N− ligands and two dmf molecules, both being centrosymmetrically (trans) related, resulting in the stoichiometric formula trans-[Co{(OPPh2)(EPPh2)N}2(dmf)2], E = S, Se. To the best of our knowledge, this is the first report of intact octahedral Co(II) complexes bearing dichalcogenidoimidodiphosphinato ligands. The previously reported octahedral [Co{(OPiPr2)(OPiPr2)NH-κ2O,O′}3]2+ species has been identified in crystals also comprising tetrahedral [Co{(OPiPr2)(OPiPr2)NH-κ2O,O′}2Cl2] and [CoCl4]2− species.48
space group and possess a center of symmetry, occupied by a Co(II) ion. The latter is coordinated to two (OPPh2)(EPPh2)N− ligands and two dmf molecules, both being centrosymmetrically (trans) related, resulting in the stoichiometric formula trans-[Co{(OPPh2)(EPPh2)N}2(dmf)2], E = S, Se. To the best of our knowledge, this is the first report of intact octahedral Co(II) complexes bearing dichalcogenidoimidodiphosphinato ligands. The previously reported octahedral [Co{(OPiPr2)(OPiPr2)NH-κ2O,O′}3]2+ species has been identified in crystals also comprising tetrahedral [Co{(OPiPr2)(OPiPr2)NH-κ2O,O′}2Cl2] and [CoCl4]2− species.48
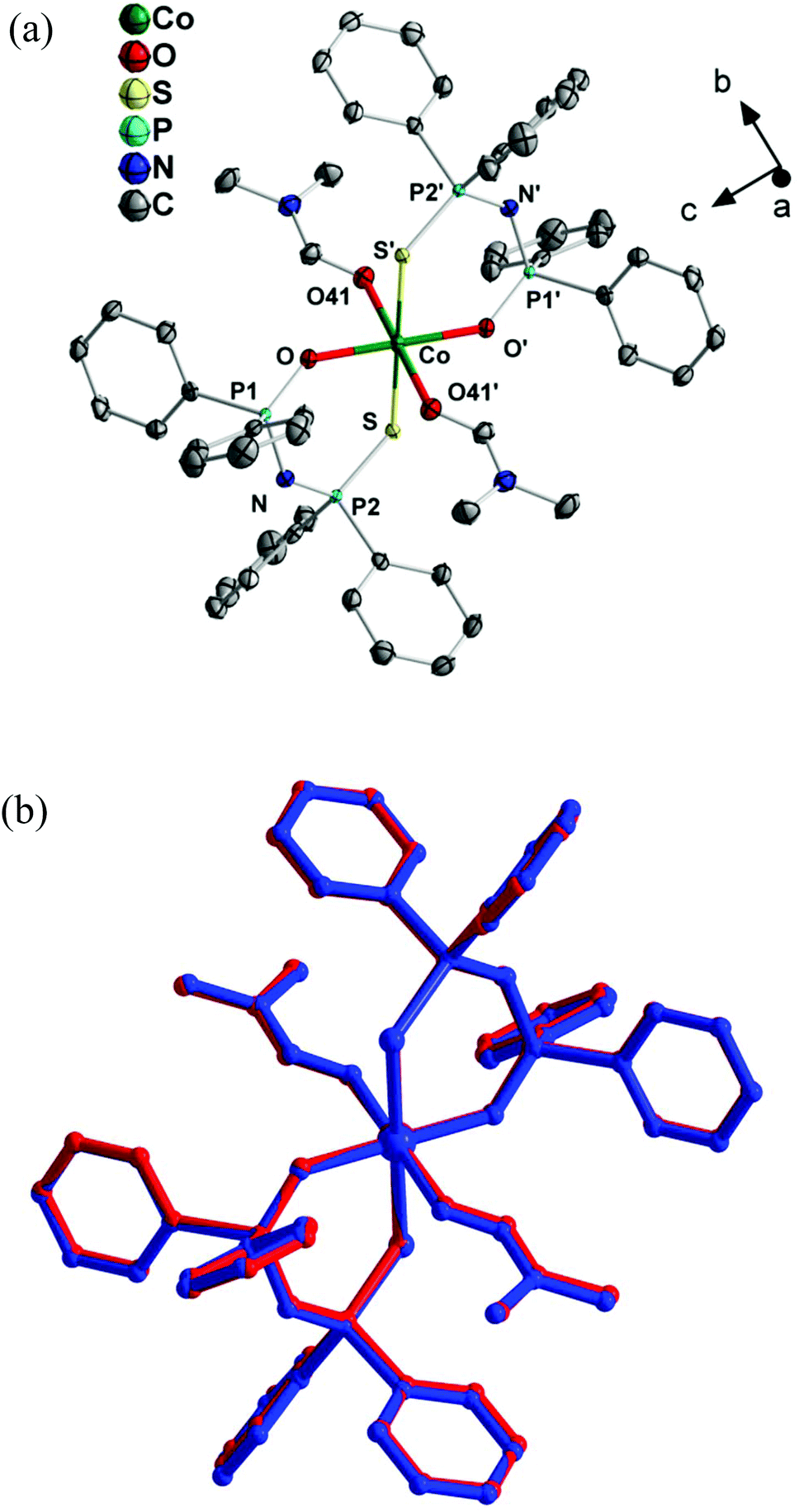 | ||
| Fig. 1 (a) Ortep plot of complex 1. Thermal ellipsoids are presented at a 50% probability level. (b) Overlay plot of the molecular structures of complexes 1 (red) and 2 (blue). Symmetry code: (′): −x, −y, −z. | ||
| 1 | 2 | ||
|---|---|---|---|
| Symmetry code: (′): −x, −y, −z. | |||
| Bond lengths (Å) | |||
| Co–O | 2.022(2) | Co–O | 2.025(2) |
| Co–O41 | 2.104(2) | Co–O41 | 2.105(2) |
| Co–S | 2.5853(8) | Co–Se | 2.6892(3) |
| Bond angles (°) | |||
| O–Co–S | 93.24(6) | O–Co–Se | 93.05(5) |
| O–Co–S′ | 86.76(6) | O–Co–Se′ | 86.95(5) |
| O41–Co–S | 90.90(6) | O41–Co–Se | 89.94(5) |
| O41–Co–S′ | 89.10(6) | O41–Co–Se′ | 90.06(5) |
| O–Co–O41 | 89.50(8) | O–Co–O41 | 89.58(7) |
| O–Co–O41′ | 90.50(8) | O–Co–O41′ | 90.42(7) |
| O41–Co–O41′ | 180.0 | O41–Co–O41′ | 180.0 |
| O–Co–O′ | 180.0 | O–Co–O′ | 180.0 |
| S–Co–S′ | 180.0 | Se–Co–Se′ | 180.0 |
The crystal structure of both 1 and 2 consists of discrete monomeric molecules, exhibiting distorted octahedral trans-CoO4E2 cores. In the following, the Co–O bond involving the chelating ligand will be referred to as Co–OL, in order to distinguish it from the Co–O41 bond involving coordination of dmf. The equatorial sites of the octahedron are occupied by four oxygen atoms with Co–OL and Co–O41 bond lengths in the range 2.022(2)–2.105(2) Å. The respective Co–OL and Co–O41 bond lengths (Fig. 1) of complexes 1 and 2 are almost equal (Table 1). The two axial sites are occupied by centrosymmetrically related S or Se atoms of the chelating ligand, with Co–E bond lengths of 2.5853(8) for 1 (E = S) and 2.6892(3) Å for 2 (E = Se), resulting in an axially elongated octahedral coordination sphere (Fig. 1 and S3†). A similar axial elongation was observed in octahedral trans-[Ni{(OPPh2)(EPPh2)N}2(dmf)2]44 and trans-[Co{(OP(OiPr)2(SCPh)N-κ2O,S}2{OP(OiPr)2(SCPh)NH-κ1O}2].49
The Co–OL bond lengths of complex 1 are larger by only 0.04 Å compared to those of tetrahedral [Co{(OPPh2)(SPPh2)N}2] (average 1.980 Å).47 By contrast, the Co–S bonds are significantly elongated by 0.25 Å and 0.27 Å/0.26 Å, compared to [Co{(OPPh2)(SPPh2)N}2] (average 2.331 Å)47 and tetrahedral [Co{(SPPh2)2N}2] (average, in two reported structures: 2.316 Å (ref. 50a) and 2.324 Å (ref. 51)), respectively. For complex 2, the Co–OL bond lengths are larger by 0.06 Å compared to those of tetrahedral [Co{(OPPh2)(SePPh2)N}2] (average 1.961 Å).44 However, the Co–Se bond lengths are larger by 0.24 Å and 0.26/0.27 Å, respectively, compared to those of [Co{(OPPh2)(SePPh2)N}2] (average 2.444 Å)44 and tetrahedral [Co{(SePPh2)2N}2] (average 2.4323![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50a and 2.415 Å (ref. 50b)). The above comparisons reveal a remarkable weakening of the Co–E bonds in complexes 1 and 2, compared to those of [Co{(EPPh2)2N}2], E = S,50a,51 Se.50 The P–N–P angles of 1 and 2 (133.0° and 133.2°, respectively) are increased slightly compared to those of the (OPPh)2(SPPh2)NH52 and {OP(OPh)2}(SePPh2)NH53 ligands (132.9° and 131.8°, respectively). The differences between the respective P–O, P–N and P–E, E = S, Se, bond lengths of these ligands and complexes 1 and 2 (not shown) are consistent with delocalization of π-electronic density over the Co–O–P–N–P–E, E = S, Se, chelate rings, upon their deprotonation and coordination to Co(II).54,55
50a and 2.415 Å (ref. 50b)). The above comparisons reveal a remarkable weakening of the Co–E bonds in complexes 1 and 2, compared to those of [Co{(EPPh2)2N}2], E = S,50a,51 Se.50 The P–N–P angles of 1 and 2 (133.0° and 133.2°, respectively) are increased slightly compared to those of the (OPPh)2(SPPh2)NH52 and {OP(OPh)2}(SePPh2)NH53 ligands (132.9° and 131.8°, respectively). The differences between the respective P–O, P–N and P–E, E = S, Se, bond lengths of these ligands and complexes 1 and 2 (not shown) are consistent with delocalization of π-electronic density over the Co–O–P–N–P–E, E = S, Se, chelate rings, upon their deprotonation and coordination to Co(II).54,55
The distortion from the ideal octahedral geometry of complexes 1 and 2 is further verified by the different endocyclic (93.24° and 93.05°, respectively) and exocyclic (86.76° and 86.95°, respectively), equatorial O–Co–E angles. Similar structural features were observed in the analogous trans-[Ni{(OPPh2)(EPPh2)N}2(dmf)2] complexes, E = S, Se, exhibiting equatorial endocyclic (93.88° and 93.27°) and exocyclic (86.12° and 86.73°) angles, respectively.44 In both 1 and 2, the two non-planar equatorial six-membered Co–O–P–N–P–E, E = S, Se, chelating rings exhibit pseudo-boat conformations, with the Co and N atoms being the “bow” and the “stern” of the boat.
DC magnetometry studies
Complexes 1 and 2 remain intact after magnetometry measurements, as deduced by elemental analysis (vide infra). Moreover, the experimental powder X-ray diffraction (PXRD) pattern of complex 1 is in good agreement with the corresponding theoretical one (Fig. S4†). For complex 2, although a good match is observed for peaks at 2θ < 10° (Fig. S5†), above this angle deviations are observed, probably caused by the coarse-grained crystalline nature of the sample, as we have not ground extensively both samples, in order to avoid their eventual damage.It should also be stressed that in analyzing magnetometric data, the employment of a spin Hamiltonian comprising Zeeman (g) and zero-field splitting terms D and E is appropriate only for orbit momentum quenched systems. In systems exhibiting either T2g or Eg ground electronic states, such a spin Hamiltonian cannot be applied.41,56,57 Unfortunately, the latter has been frequently employed for octahedral Co(II) complexes, affording the D and E data listed in Table S1,† but, owing to the above discrepancy, these data should be treated very cautiously.
The temperature-dependent magnetic susceptibility of complexes 1 and 2 was measured under a DC field of 1000 Oe, in the temperature range between 2 and 300 K (Fig. 2a). The χmT value at 300 K is 3.27 emu mol−1 K for 1 and 3.60 emu mol−1 K for 2, which are much larger than the theoretical value of 1.875 emu mol−1 K for a spin-only high spin Co(II) system.58 It is worth noting the strong temperature dependence of χmT which is different from the typical spin-only magnetic anisotropy behaviour. These data suggest the existence of significant SOC of the spin carriers.
 | ||
| Fig. 2 (a) Temperature-dependent χmT data for 1 and 2 between 2 and 300 K under a DC field of 1000 Oe. (b, c) M vs. H/T plots for 1 and 2, respectively, at low temperature values. | ||
The low-temperature magnetization of 1 and 2 was measured at different DC fields below 4 T. The nearly superposition of the M vs. H/T plots (Fig. 2b and c) implies that the magnetic field is too small to vary the population of the electronic fine structures at low temperature. This is strong evidence for the existence of significant magnetic anisotropy in complexes 1 and 2. However, owing to the significant contribution of the orbital angular momentum in octahedral complexes 1 and 2, the ANISOFIT 2.0 software59 is not suitable to fit the data. To describe the DC magnetic properties of these systems, the Griffith Hamiltonian (1) was employed, which includes the Δax and Δrh crystal field parameters (vide supra) and explicitly takes into account the unquenched orbital angular momentum of the Co(II) ion:60–62
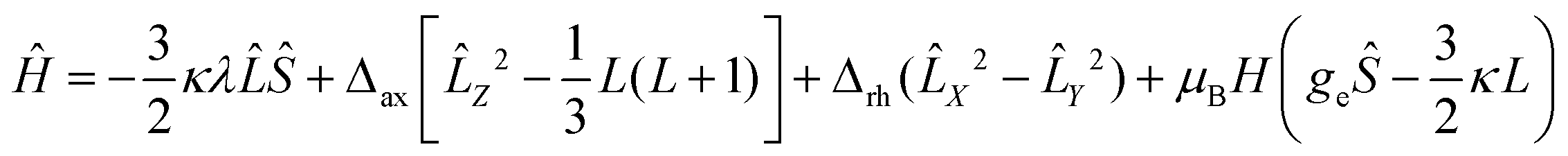 | (1) |
In eqn (1), λ is the spin–orbit coupling parameter and κ is the reduction factor of the orbital angular momentum. The fitting results were obtained through the use of the PHI software.63 By setting κ = −1.3 and fixing gx = gy = 2 (implying axial magnetic anisotropy, vide infra), the parameters listed in Table 2 were obtained. Both complexes 1 and 2 exhibit very small Δrh values and, therefore, an axial type of magnetic anisotropy. Moreover, the fact that in both systems Δax < 0 reveals the existence of an easy axis of magnetization.
| Complex | g z | Δax (cm−1) | Δrh (cm−1) | λ (cm−1) | U eff (cm−1) |
|---|---|---|---|---|---|
| 1 | 2.40 | −487.4 | 0.15 | −138.6 | 23.6 |
| 2 | 2.78 | −448.4 | −0.43 | −153.3 | 18.1 |
Angular-dependent magnetometry studies of complex 1
A suitably large crystal of complex 1 was oriented as shown in Fig. S6† and studied by magnetometry. Strong anisotropy of the magnetization was observed for the rotations along experimentally defined X- (Fig. 3a) and Z- (Fig. 3c) directions, whereas along the Y-direction (Fig. 3b) the magnetic susceptibility was found to be more isotropic and having a smaller overall value. This observation provides strong evidence of axial anisotropy and the presence of an easy axis of magnetization, confirming the data obtained by DC magnetometry (vide supra). The susceptibility tensor with respect to the experimental frame is determined by simultaneously fitting the three rotation sine curves at 3 K, as depicted in Fig. 3d. The principle axes and the corresponding susceptibility values are therefore the eigenvectors and eigenvalues of the tensor. As shown in Fig. 4a, the χmT value along the easy axis is larger than those along the other two axes, thus unequivocally confirming that complex 1 exhibits axial anisotropy and possesses an easy axis of magnetization. The orientation of the experimentally determined magnetization principal axes, with respect to the XYZ frame (Fig. S6†), is shown in Fig. 4b. Attempts were made to carry out a similar study on a crystal of complex 2, but its smaller size, compared to that of complex 1, precluded such experiments.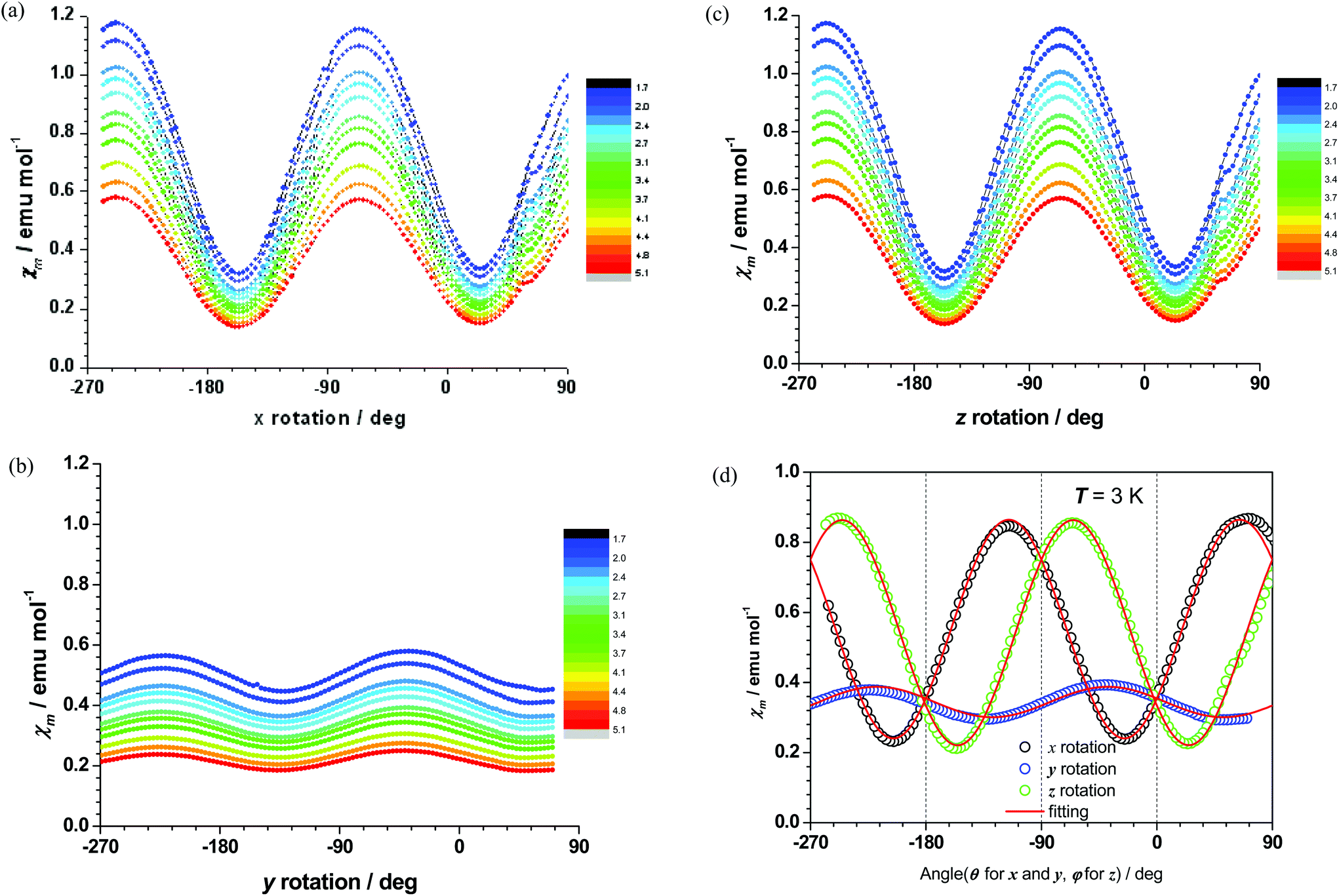 | ||
| Fig. 3 (a, b, c, respectively) The magnetic susceptibility of complex 1 along the X, Y, Z directions between 1.8 K and 5 K. (d) Angular dependence of the magnetic susceptibility of 1 at 3 K under a DC field of 1000 Oe (circle). The red solid lines represent the best fitting based on the experimental data. | ||
 | ||
| Fig. 4 (a) The χmT values of complex 1 determined by angular-dependent magnetometry measurements along the easy (red), medium (blue) and hard (black) axes. (b) Experimentally determined magnetization principal axes, with the red arrow being the easy axis. | ||
AC magnetometry studies
To probe the dynamic magnetic behavior of complexes 1 and 2, their AC magnetic susceptibility was measured at various frequencies and temperatures. Under zero-applied DC magnetic field, no slow relaxation of magnetization was observed in the 10–1000 Hz and 100–10000 Hz range for 1 and 2, respectively (Fig. S7a and d†). These observations provide evidence of a fast quantum tunneling of magnetization (QTM), which is stemming from extensive mixing of excited states into the ground states.64,65 On the other hand, both complexes 1 and 2 exhibit slow magnetic relaxations under an external DC field (300 and 2000 Oe, respectively), which can prominently suppress the QTM process (Fig. S7b, c and e†). It should be noted that, to the best of our knowledge, all complexes listed in Table S1† exhibit slow relaxation of magnetization only in the presence of an external DC magnetic field.
The relaxation times τ of complexes 1 and 2 were extracted by fitting χ′ and χ′′ to a generalized Debye model. The corresponding Cole–Cole plots for complexes 1 and 2 are shown in Fig. S8 and S9,† respectively. The distribution of the corresponding α values is very narrow, indicating a single relaxation process. By fitting lnτ vs. T−1 on the basis of an Arrhenius expression (Fig. 5c and 6b), effective energy barriers Ueff = 23.6 cm−1 for 1 (under 300 Oe) and Ueff = 18.1 cm−1 for 2 (under 2000 Oe) were obtained. For complex 2, due to the curvature of the plot, in addition to the Arrhenius-type fit performed at the high temperature region, a fit according to the equation τ−1 = CTn for a Raman relaxation process was carried out (Fig. 6b). We note that the Ueff values of complexes 1 and 2 are significantly smaller compared to the corresponding Δax parameters of the two complexes (Table 2) and fall between the extremes of values corresponding to the Co(II) octahedral complexes listed in Table S1.†
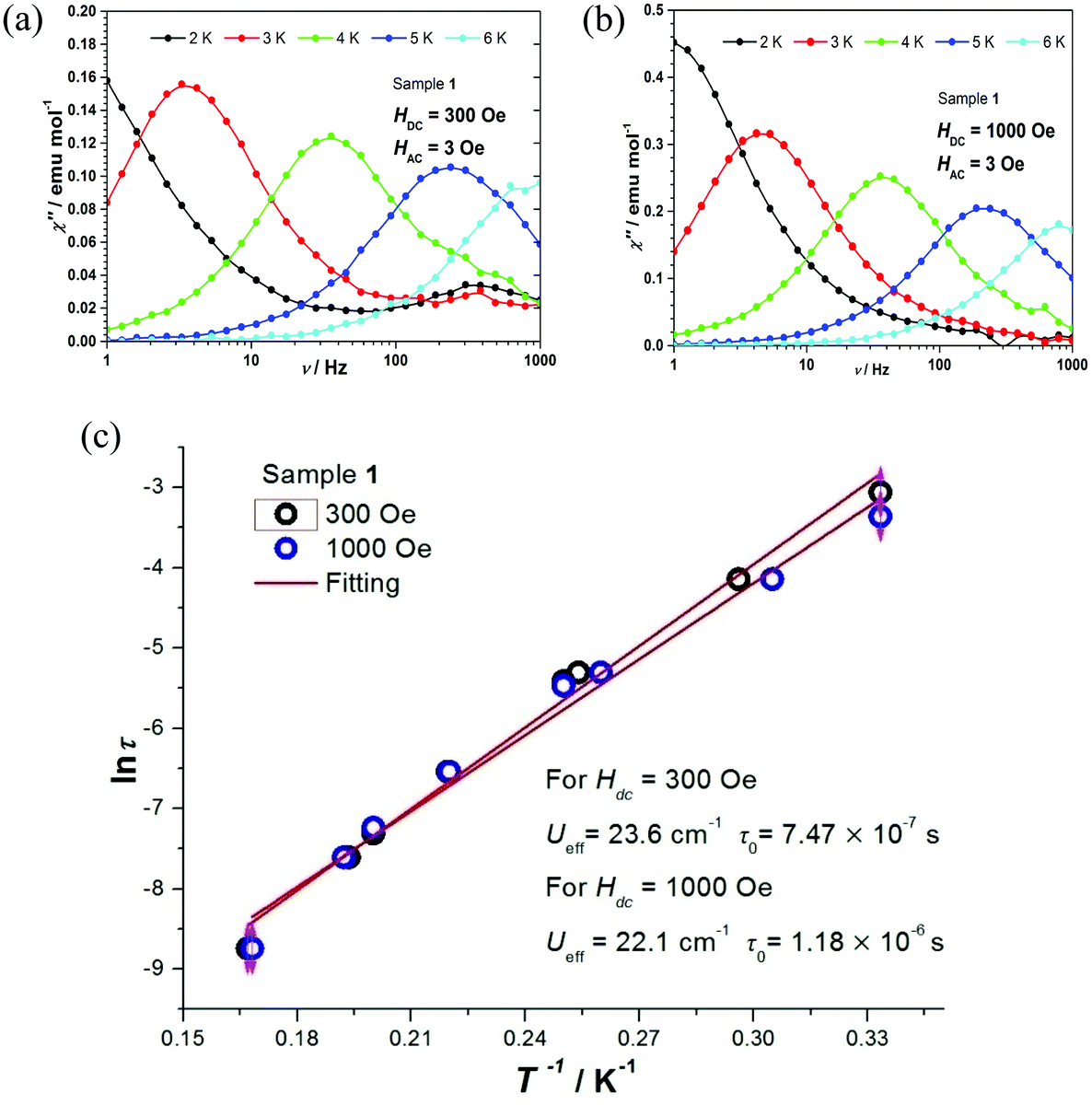 | ||
| Fig. 5 Complex 1: Frequency dependence of the out-of-phase AC susceptibility under 300 Oe (a) and 1000 Oe (b) from 2 K to 6 K. (c) lnτ vs. T−1 under 300 Oe (black circles) and 1000 Oe (blue circles). Red lines represent Arrhenius fit plots. | ||
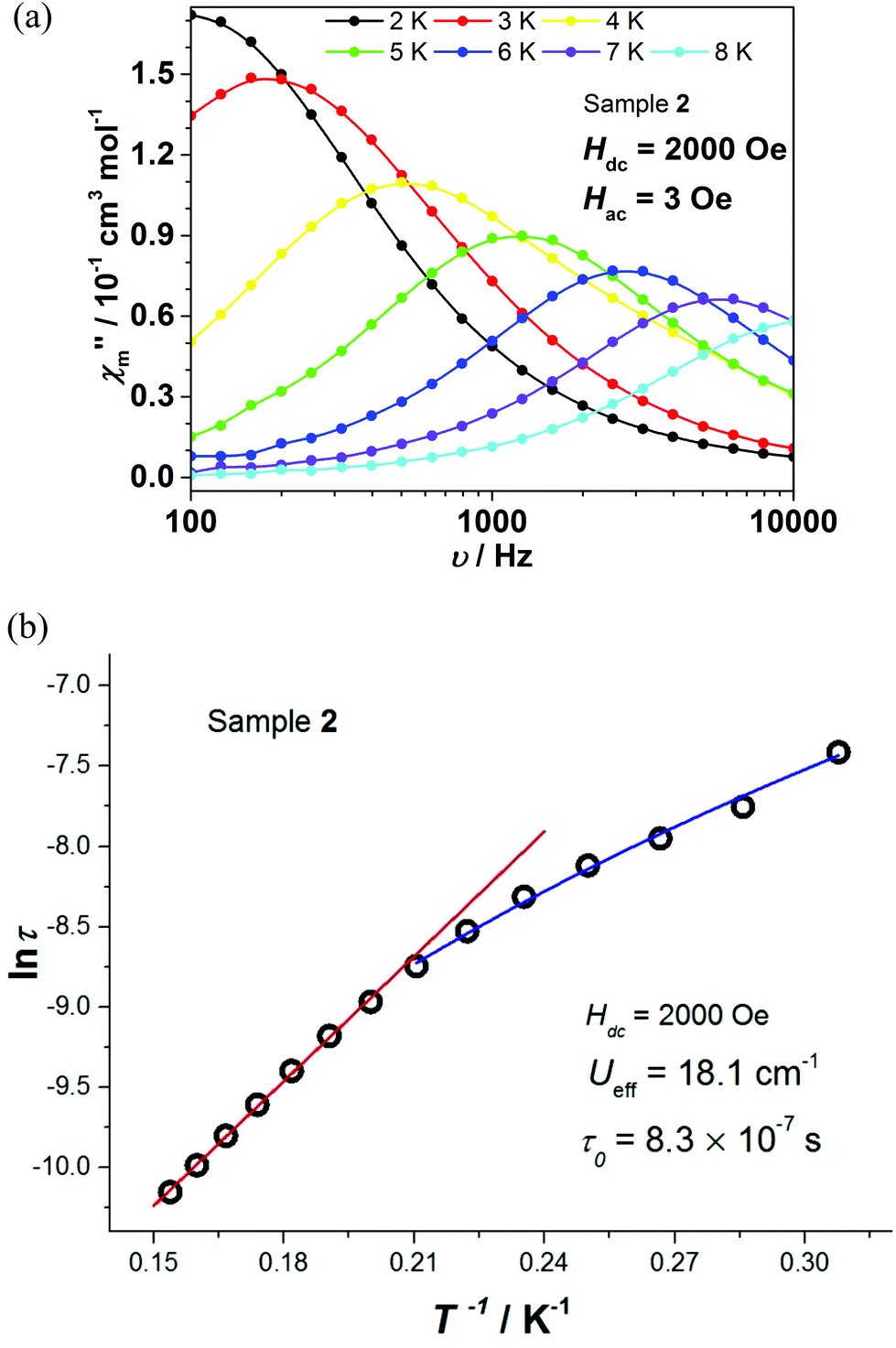 | ||
| Fig. 6 Complex 2: (a) Frequency dependence of the out-of-phase AC susceptibility under 2000 Oe from 2 K to 8 K. (b) Arrhenius-type lnτ vs. T−1 fit (high temperature region, red line) and Raman relaxation fit τ−1 = CTn (low temperature region, blue line, with fitting parameters C = 30.4 and n = 3.4), under 2000 Oe. | ||
Effects of S/Se metal-coordination
This work reports on the magnetic anisotropy and relaxation properties of octahedral complexes bearing Co–E, E = S, Se, bonds. The Δax and Δrh values of complexes 1 and 2 reveal that replacing S in 1 by Se in 2 leads to smaller axial and larger rhombic anisotropy (Table 2). The smaller magnitude of Δax in complex 2 may be caused by the greater degree of covalency of the Co–Se bond compared to that of the Co–S bond, leading to stronger quenching of the metal ion's SOC, due to the ligand field, in complex 2.22 On the other hand, the magnetic anisotropy is also expected to be affected by the nature of the ligands. For instance, in some systems, heavier donor atoms have been shown to confer stronger axial anisotropy, most likely due to their stronger SOC (vide infra). However, the respective operating effects cannot be quantified in a straightforward manner, because the local coordination geometry of the metal ions, as well as the metal–ligand covalency, may vary among different systems.A similar trend, to that established for complexes 1 and 2, has been previously observed concerning trans-[Ni{(OPPh2)(EPPh2)N}2(sol)2], E = S, Se, for sol = dmf, thf, in which the E = Se complex exhibits smaller D but larger E/D.45 On the other hand, both smaller D and E/D were observed for the Se-containing complex when sol = dmso, as determined by HFEPR.46 It should be also noted that the tetrahedral S = 3/2 [Co{(EPiPr2)(EiPr2)N}2] complexes, exhibit similar experimentally determined axial anisotropies (D ∼ −30.5, E = S and −30.4 cm−1 E = Se),38 whereas the E = Te analogue shows larger axial anisotropy (D = −45.1 cm−1).39 No significant differences have been observed in the magnetic anisotropy between tetrahedral [Co{(EPPh2)2N}2], E = S (−11.9 cm−1),66 Se (−15.8 cm−1).39 On the other hand, a trend of progressively larger axial anisotropies was established for tetrahedral [Co(EPh)4]2− complexes along the E = O, S, Se, series.67 Furthermore, in the case of the tetrahedral S = 2 complexes [Fe{(EPPh2)2N}2], the E = Se complex68 exhibits similar axial but slightly larger rhombic anisotropy, compared with that of the E = S one,69 as determined by HFEPR. Unlike the moderate effects, on the magnetic anisotropy, of Se- versus S-coordination to the above 3d metal ions, the presence of heavier p-block elements like Se enhances the magnetic anisotropy of S = ½ radicals, due to increased SOC effects.70,71
In the above context, the coordination of heavier halogeno ligands to Ni(II)72 or Cr(II)73 has been shown to confer increased axial anisotropies. Similar effects, on the magnitude of the axial anisotropy due to the coordination of heavier halides, have also been established in tetrahedral74–80 Co(II) complexes, in some instances imposing a concomitant change in the sign of D as well.76,77 On the other hand, contrary to the above observations, lower axial anisotropy due to the coordination of heavier halides has been observed in square-pyramidal81,82 and pentagonal bipyramidal83 Co(II) complexes.
With respect to the dynamic magnetic properties, complex 2, bearing a CoO4Se2 coordination sphere, exhibits smaller Ueff energy barrier (18.1 cm−1) than that of complex 1 (23.6 cm−1) bearing a CoO4S2 one (Tables 2 and S1†). This observation is compatible with the fact that complex 2 exhibits smaller axial and larger rhombic anisotropy compared with that of complex 1 (vide supra). In the same context, the Ueff values and relaxation mechanisms for the series of tetrahedral S = 3/2 [Co{(EPiPr2)2N}2] complexes, E = S (54 cm−1, at 0.2 Tesla, Orbach mechanism),38 Se (Raman mechanism, at zero-field),38 Te (22 cm−1, Orbach mechanism, at zero-field),39 reveal profound effects of the nature of the donor atom E. On the other hand, the Ueff values for the series of tetrahedral [Co(EPh)4]2− complexes, E = O (21 cm−1), S (21 cm−1), Se (19 cm−1), are effectively the same.67
Owing to the above observations that do not establish clear trends, additional experimental and computation work is needed in order to fully elucidate the effects, on either magnetic anisotropy or relaxation properties, of metal–ligand coordination via heavier donor atoms.
Conclusions
The synthesis and characterization of trans-[Co{(OPPh2)(EPPh2)N}2(dmf)2], E = S, Se, complexes is described, based on the crystallization of tetrahedral [Co{(OPPh2)(EPPh2)N}2] in the presence of dmf. X-ray crystallography studies revealed that complexes 1 and 2 are isomorphous and they exhibit axially elongated octahedral trans-CoO4E2, E = S, Se, coordination spheres. DC magnetometry analysis established that both complexes exhibit axial magnetic anisotropy, which, for complex 1, was unequivocally confirmed by magnetometry studies on an oriented crystal of this complex, also revealing an easy axis of magnetization.This work extends the dataset of octahedral Co(II) complexes exhibiting field-induced slow relaxation of magnetization, which are dominated by the presence of Co–O or Co–N bonds in their coordination spheres (Table S1†). Moreover, AC magnetometry studies probed the dynamic magnetic properties of the two complexes, revealing an apparently smaller Ueff value for the Se-containing complex 2 compared with that of the S-containing complex 1 (Tables 2 and S1†). Complexes 1 and 2 extend the set of 3d-based complexes bearing dichalcogenidoimidodiphosphinato ligands40 and exhibiting slow relaxation of magnetization, so far consisting of tetrahedral Co(II)38,39 and octahedral Mn(III) complexes.84
Materials and methods
General
The ligands (OPPh2)(EPPh2)NH, E = S,52 Se,52 and complexes [Co{(OPPh2)(EPPh2)N}2] E = S,47 Se,44 were prepared according to published procedures. Elemental analyses were performed prior and after magnetometry measurements using Elementar Vario MICRO CUBE (Germany). IR spectra were run in the range 4000–200 cm−1 on a PerkinElmer 883 IR spectrophotometer, as KBr pellets. The PXRD patterns were recorded on a Rigaku R-AXIS IV Imaging Plate Detector mounted on a Rigaku RU-H3R Rotating Copper Anode X-ray Generator (λ = 1.54 Å). The samples were slightly ground, inserted in capillary mark tubes (ID = 1 mm) and attached to a rotating stage. The capillaries were spinned (36° min−1) during the measurement.Chemical synthesis
DC magnetic susceptibility measurements for complex 2 were performed on a Quantum Design MPMSXL5 SQUID system. AC susceptibility for 2 was measured on a Quantum Design PPMS-9 with ACMS accessories in the 100–10000 Hz.
Angular-dependent magnetometric studies on a single crystal of complex 1 were carried out on the MPMSXL7 SQUID magnetometer equipped with a horizontal rotator. A single crystal of 1 (0.48 mg) was mounted with its (100) face glued on an L-shaped Cu/Be support, in order to perform a rotation around the support's three orthogonal axes. The orientation of this crystal for magnetometry studies is described in Fig. S6.† Three series rotation data were collected under temperatures between 1.8 K and 5 K, at 10000 Oe, and subsequently corrected for the diamagnetic contribution of the support and grease. The detailed experimental procedure has been described in the literature.88
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
P. K. would like to thank the Special Account of the National and Kapodistrian University of Athens for financial support. E. F. has been awarded a State Scholarship Foundation (IKY) fellowship, by the act “Support for Postdoctoral Researchers”, from the operational program “Human Power Development, Education and Lifelong Learning” (6, 8, 9 priority axes), co-funded by the European Social Fund and the Greek State. V. P. would like to thank the Special Account of NCSR “Demokritos” for financial support concerning the operation of the X-ray facilities at INN through the internal program entitled “Structural study and characterization of crystalline materials” (NCSR Demokritos, ELKE #10 813). S.-D. J. thanks the National Natural Science Foundation of China (21822301), the National Basic Research Program of China (2018YFA0306003) and the Beijing National Laboratory for Molecular Sciences (Y18G23), for funding. We are grateful to Dr Theodore Steriotis (INN, NCSR “Demokritos”) for his contribution to the PXRD measurements, as well as to the Reviewers of the manuscript for their constructive comments.References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS.
- D. Gatteschi, L. Bogani, A. Cornia, M. Mannini, L. Sorace and R. Sessoli, Solid State Sci., 2008, 10, 1701–1709 CrossRef CAS.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- J. W. Sharples and D. Collison, Polyhedron, 2013, 66, 15–27 CrossRef CAS.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- G. Aromi and E. K. Brechin, Struct. Bonding, 2006, 122, 1–67 CrossRef CAS.
- C. J. Milios and R. E. P. Winpenny, in Molecular Nanomagnets and Related Phenomena, ed. S. Gao, 2015, vol. 164, pp. 1–109 Search PubMed.
- C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 2754–2755 CrossRef CAS PubMed.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clerac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926–4929 CrossRef CAS PubMed.
- J. Cirera, E. Ruiz, S. Alvarez, F. Neese and J. Kortus, Chem. – Eur. J., 2009, 15, 4078–4087 CrossRef CAS PubMed.
- F. Neese and D. A. Pantazis, Faraday Discuss., 2010, 148, 229–238 RSC.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- Y. S. Meng, S.-D. Jiang, B. W. Wang and S. Gao, Acc. Chem. Res., 2016, 49, 2381–2389 CrossRef CAS PubMed.
- K. R. Meihaus and J. R. Long, Dalton Trans., 2015, 44, 2517–2528 RSC.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 1062, 1400–1403 CrossRef PubMed.
- K. Randall McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 8492–8503 RSC.
- G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- A. K. Bar, C. Pichon and J. P. Sutter, Coord. Chem. Rev., 2016, 308, 346–380 CrossRef CAS.
- M. Feng and M. L. Tong, Chem. – Eur. J., 2018, 24, 7574–7594 CrossRef CAS PubMed.
- S. Gomez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289, 379–392 CrossRef.
- M. Atanasov, D. Aravena, E. Suturina, E. Bill, D. Maganas and F. Neese, Coord. Chem. Rev., 2015, 289, 177–214 CrossRef.
- D. E. Freedman, W. H. Harman, T. D. Harris, G. J. Long, C. J. Chang and J. R. Long, J. Am. Chem. Soc., 2010, 132, 1224–1225 CrossRef CAS PubMed.
- M. Atzori, L. Tesi, E. Morra, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 2154–2157 CrossRef CAS PubMed.
- M. Atzori, S. Benci, E. Morra, L. Tesi, M. Chiesa, R. Torre, L. Sorace and R. Sessoli, Inorg. Chem., 2018, 57, 731–740 CrossRef CAS PubMed.
- C. Rajnák, J. Titiš, J. Moncol, R. Mičová and R. Boča, Inorg. Chem., 2019, 58, 991–994 CrossRef PubMed.
- A. C. Benniston, S. Melnic, C. Turta, A. B. Arauzo, J. Bartolomé, E. Bartolomé, R. W. Harrington and M. R. Probert, Dalton Trans., 2014, 43, 13349–13357 RSC.
- S. Q. Wu, Y. Miyazaki, M. Nakano, S. Q. Su, Z. S. Yao, H. Z. Kou and O. Sato, Chem. – Eur. J., 2017, 23, 10028–10033 CrossRef CAS PubMed.
- S. Tripathi, A. Dey, M. Shanmugam, R. S. Narayanan and V. Chandrsekhar, in Topics in Organometallic Chemistry, Springer, 2018, DOI:10.1007/3418_2018_8.
- D. Swenson, N. C. Baenziger and D. Coucouvanis, J. Am. Chem. Soc., 1978, 100, 1932–1934 CrossRef CAS.
- K. Fukui, N. Kojima, H. Ohya-Nishiguchi and N. Hirota, Inorg. Chem., 1992, 31, 1338–1344 CrossRef CAS.
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS PubMed.
- E. A. Suturina, D. Maganas, E. Bill, M. Atanasov and F. Neese, Inorg. Chem., 2015, 54, 9948–9961 CrossRef CAS PubMed.
- E. A. Suturina, J. Nehrkorn, J. M. Zadrozny, J. Liu, M. Atanasov, T. Weyhermuller, D. Maganas, S. Hill, A. Schnegg, E. Bill, J. R. Long and F. Neese, Inorg. Chem., 2017, 56, 3102–3118 CrossRef CAS PubMed.
- M. Craven, M. H. Nygaard, J. M. Zadrozny, J. R. Long and J. Overgaard, Inorg. Chem., 2018, 57, 6913–6920 CrossRef CAS PubMed.
- S. Sottini, G. Poneti, S. Ciattini, N. Levesanos, E. Ferentinos, J. Krzystek, L. Sorace and P. Kyritsis, Inorg. Chem., 2016, 55, 9537–9548 CrossRef CAS PubMed.
- X. N. Yao, M. W. Yang, J. Xiong, J. J. Liu, C. Gao, Y. S. Meng, S.-D. Jiang, B. W. Wang and S. Gao, Inorg. Chem. Front., 2017, 4, 701–705 RSC.
- I. Haiduc, Dichalcogenoimidodiphosphinato Ligands in Comprehensive Coordination Chemistry II: From Biology to Nanotechnology, ed. J. A. McCleverty and T. J. Meyer, in Fundamentals, ed. A. B. P. Lever, Elsevier, Amsterdam, 2004, vol. 1, pp. 323–347 Search PubMed.
- J. Titiš and R. Boča, Inorg. Chem., 2011, 50, 11838–11845 CrossRef PubMed.
- J. Vallejo, I. Castro, R. Ruiz-Garcia, J. Cano, M. Julve, F. Lloret, G. De Munno, W. Wernsdorfer and E. Pardo, J. Am. Chem. Soc., 2012, 134, 15704–15707 CrossRef CAS PubMed.
- A. Silvestru, D. Bilc, R. Rosler, J. E. Drake and I. Haiduc, Inorg. Chim. Acta, 2000, 305, 106–110 CrossRef CAS.
- E. Ferentinos, D. Maganas, C. P. Raptopoulou, A. Terzis, V. Psycharis, N. Robertson and P. Kyritsis, Dalton Trans., 2011, 40, 169–180 RSC.
- D. Maganas, J. Krzystek, E. Ferentinos, A. M. Whyte, N. Robertson, V. Psycharis, A. Terzis, F. Neese and P. Kyritsis, Inorg. Chem., 2012, 51, 7218–7231 CrossRef CAS PubMed.
- E. Ferentinos, C. P. Raptopoulou, V. Psycharis, A. Terzis, J. Krzystek and P. Kyritsis, Polyhedron, 2018, 151, 177–184 CrossRef CAS.
- M. C. Aragoni, M. Arca, M. B. Carrea, A. Garau, F. A. Devillanova, F. Isaia, V. Lippolis, G. L. Abbati, F. Demartin, C. Silvestru, S. Demeshko and F. Meyer, Eur. J. Inorg. Chem., 2007, 4607–4614 CrossRef CAS.
- N. Levesanos, A. Grigoropoulos, C. P. Raptopoulou, V. Psycharis and P. Kyritsis, Inorg. Chem. Commun., 2013, 30, 34–38 CrossRef CAS.
- F. D. Sokolov, D. A. Safin, N. G. Zabirov, L. N. Yamalieva, D. B. Krivolapov and I. A. Litvinov, Mendeleev Commun., 2004, 14, 51–52 CrossRef.
- (a) L. M. Gilby and B. Piggott, Polyhedron, 1999, 18, 1077–1082 CrossRef CAS; (b) J. Novosad, M. Necas, J. Marek, P. Veltsistas, C. Papadimitriou, I. Haiduc, M. Watanabe and J. D. Woollins, Inorg. Chim. Acta, 1999, 290, 256–260 CrossRef CAS.
- M. C. Aragoni, M. Arca, A. Garau, F. Isaia, V. Lippolis, G. L. Abbati and A. C. Fabretti, Z. Anorg. Allg. Chem., 2000, 626, 1454–1459 CrossRef CAS.
- A. M. Z. Slawin, M. B. Smith and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1996, 3659–3665 RSC.
- M. Necas, M. R. S. Foreman, J. Marek, J. D. Woollins and J. Novosad, New J. Chem., 2001, 25, 1256–1263 RSC.
- C. Silvestru and J. E. Drake, Coord. Chem. Rev., 2001, 223, 117–216 CrossRef CAS.
- D. Maganas, S. S. Staniland, A. Grigoropoulos, F. White, S. Parsons, N. Robertson, P. Kyritsis and G. Pneumatikakis, Dalton Trans., 2006, 2301–2315 RSC.
- D. V. Korchagin, A. V. Palii, E. A. Yureva, A. V. Akimov, E. Y. Misochko, G. V. Shilov, A. D. Talantsev, R. B. Morgunov, A. A. Shakin, S. M. Aldoshin and B. S. Tsukerblat, Dalton Trans., 2017, 46, 7540–7548 RSC.
- R. Boča, C. Rajnák, J. Moncol, J. Titiš and D. Valigura, Inorg. Chem., 2018, 57, 14314–14321 CrossRef PubMed.
- J. R. Pilbrow, J. Magn. Reson., 1978, 31, 479–490 CAS.
- M. P. Shores, J. J. Sokol and J. R. Long, J. Am. Chem. Soc., 2002, 124, 2279–2292 CrossRef CAS PubMed.
- J. S. Griffith, The Theory of Transition Metal Ions, University Press, Cambridge, 1964 Search PubMed.
- O. Kahn, Molecular Magnetism, VCH Publishers, New York, 1993 Search PubMed.
- F. Lloret, M. Julve, J. Cano, R. Ruiz-García and E. Pardo, Inorg. Chim. Acta, 2008, 361, 3432–3445 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, U.K., 2006 Search PubMed.
- W. Wernsdorfer and R. Sessoli, Science, 1999, 284, 133–135 CrossRef CAS PubMed.
- D. Maganas, S. Milikisyants, J. M. A. Rijnbeek, S. Sottini, N. Levesanos, P. Kyritsis and E. J. J. Groenen, Inorg. Chem., 2010, 49, 595–605 CrossRef CAS PubMed.
- J. M. Zadrozny, J. Telser and J. R. Long, Polyhedron, 2013, 64, 209–217 CrossRef CAS.
- E. Ferentinos, S. Chatziefthimiou, A. K. Boudalis, M. Pissas, G. Mathies, P. Gast, E. J. J. Groenen, Y. Sanakis and P. Kyritsis, Eur. J. Inorg. Chem., 2018, 713–721 CrossRef CAS.
- G. Mathies, S. D. Chatziefthimiou, D. Maganas, Y. Sanakis, S. Sottini, P. Kyritsis and E. J. J. Groenen, J. Magn. Reson., 2012, 224, 94–100 CrossRef CAS PubMed.
- S. M. Winter, S. Hill and R. T. Oakley, J. Am. Chem. Soc., 2015, 137, 3720–3730 CrossRef CAS PubMed.
- S. M. Winter, S. Datta, S. Hill and R. T. Oakley, J. Am. Chem. Soc., 2011, 133, 8126–8129 CrossRef CAS PubMed.
- J. Krzystek, J.-H. Park, M. W. Meisel, M. A. Hitchman, H. Stratemeier, L.-C. Brunel and J. Telser, Inorg. Chem., 2002, 41, 4478–4487 CrossRef CAS PubMed.
- H. I. Karunadasa, K. D. Arquero, L. A. Berben and J. R. Long, Inorg. Chem., 2010, 49, 4738–4740 CrossRef CAS PubMed.
- M. R. Saber and K. R. Dunbar, Chem. Commun., 2014, 12266–12269 RSC.
- L. Smolko, J. Cernak, J. Kuchar, C. Rajnak, J. Titis and R. Boca, Eur. J. Inorg. Chem., 2017, 3080–3086 CrossRef CAS.
- S. Vaidya, S. K. Singh, P. Shukla, K. Ansari, G. Rajaraman and M. Shanmugam, Chem. – Eur. J., 2017, 23, 9546–9559 CrossRef CAS PubMed.
- S. Vaidya, A. Upadhyay, S. K. Singh, T. Gupta, S. Tewary, S. K. Langley, J. P. S. Walsh, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. Commun., 2015, 51, 3739–3742 RSC.
- L. Smolko, J. Cernak, M. Dusek, J. Titis and R. Boca, New J. Chem., 2016, 40, 6593–6598 RSC.
- A. K. Mondal, M. Sundararajan and S. Konar, Dalton Trans., 2018, 47, 3745–3754 RSC.
- D. V. Korchagin, G. V. Shilov, S. M. Aldoshin, R. B. Morgunov, A. D. Talantsev and E. A. Yureva, Polyhedron, 2015, 102, 147–151 CrossRef CAS.
- J. J. Liu, Y. S. Meng, I. Hlavicka, M. Orlita, S.-D. Jiang, B. W. Wang and S. Gao, Dalton Trans., 2017, 46, 7408–7411 RSC.
- A. K. Mondal, T. Goswami, A. Misra and S. Konar, Inorg. Chem., 2017, 56, 6870–6878 CrossRef CAS PubMed.
- B. Drahos, R. Herchel and Z. Travnicek, Inorg. Chem., 2017, 56, 5076–5088 CrossRef CAS PubMed.
- A. Grigoropoulos, M. Pissas, P. Papatolis, V. Psycharis, P. Kyritsis and Y. Sanakis, Inorg. Chem., 2013, 52, 12869–12871 CrossRef CAS PubMed.
- CrystalClear, Rigaku/MSC Inc., The Woodlands, Texas, USA, 2005 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- DIAMOND – Crystal and Molecular Structure Visualization, Ver. 3.1, Crystal Impact, Rathausgasse 30, 53111, Bonn, Germany Search PubMed.
- S.-D. Jiang, B. W. Wang and S. Gao, in Molecular Nanomagnets and Related Phenomena, ed. S. Gao, 2015, vol. 164, pp. 111–141 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Complexes 1 and 2: crystal data and crystallographic information files, orientation of a crystal of 1, IR spectroscopy, PXRD and magnetometry data for 1 and 2, literature survey on slowly-relaxing octahedral Co(II) complexes. CCDC 1892040 and 1892041. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi00135b |
| ‡ These authors contributed equally. |
| § Present address: Department of Chemistry, Materials Innovation Factory, University of Liverpool, Liverpool L7 3NY, U.K. |
| This journal is © the Partner Organisations 2019 |