
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular-salt hybrids; integration of ammonia borane into lithium halides†
Irene
Cascallana-Matías
 ab,
Joachim
Breternitz‡
a,
Annabelle
Baker
c,
Hallam
Davis
a,
Edmund J.
Cussen§
b and
Duncan H.
Gregory
*a
ab,
Joachim
Breternitz‡
a,
Annabelle
Baker
c,
Hallam
Davis
a,
Edmund J.
Cussen§
b and
Duncan H.
Gregory
*a
aWestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Duncan.Gregory@Glasgow.ac.uk
bWestCHEM, Dept of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK
cDiamond Light Source, Harwell Oxford, Didcot, Oxfordshire OX11 0QX, UK
First published on 18th February 2019
Abstract
New molecular-salt-like hybrids contain ammonia borane (AB) in co-existence with lithium halides in single lattices. [LiI](NH3BH3) and [LiI](NH3BH3)2 are members of a generic [LiX]m(AB)n (X = I−, BH4−; m, n = integer) materials family which release hydrogen on decomposition and demonstrate Li+-ion conductivity more than two orders of magnitude higher than either LiBH4 or LiI.
Light metal complex hydrides such as lithium alanates and borohydrides have attracted immense interest given the high values of pseudo-reversible gravimetric hydrogen capacity that are possible.1–4 Moreover, the decomposition and release of hydrogen from a number of complex hydrides such as lithium borohydride is linked to a transition to a fast lithium ion conducting state.5,6 LiBH4 has an inherently low Li+ ion conductivity under ambient conditions that can be improved via phase transitions that occur at higher temperature (HT) or pressure (HP). The HT polymorph can be stabilised at room temperature by partial replacement of (BH4)− with Br− or I−, leading to an increase in conductivity by several orders of magnitude at 30 °C.7,8 Other hydrides of even higher complexity such as amide borohydrides can exhibit Li ion transport into the superionic regime with values of conductivity at ambient and elevated temperatures far exceeding those of the equivalent component binary lithium compounds themselves.9,10 In fact, from a practical standpoint, complex borohydrides exhibit surprisingly good compatibility with Li (or Na) electrodes and a high electrochemical stability.5 Moreover, the electrolyte–electrode interface is not difficult to fabricate, simplifying manufacture of battery architectures, although coatings between both components are required.6
Similar to the complex hydrides, the molecular hydride, ammonia borane (AB), NH3BH3, has long been considered as a promising candidate for high capacity hydrogen storage applications.11–14 AB has a low molecular weight (30.7 g mol−1), satisfactory air-stability and is thermally stable above 100 °C. Several lithium aminoboranes have been characterised in terms of their hydrogen storage properties,15 but unlike the borohydrides, their Li-ion conductivity is unknown.
Here, we investigate the Li-ion transport properties of molecular hydride-metal hydride/halide materials for the first time. The design of these solids incorporates molecular and ionic components in hybrid structures and as part of this study we report the Li-ion transport properties of the ammonia borane borohydride, [LiBH4]2NH3BH3 and the related lithium diamidoborane borohydride, Li(BH3NH2BH2NH2BH3); two compounds formerly considered as hydrogen storage materials. Moreover, by following a hybrid materials design strategy, we demonstrate that it is possible to synthesise two novel lithium ammonia borane iodides, LiI[NH3BH3] and LiI[NH3BH3]2. The structure, thermal stability and decomposition behaviour of the compounds was elucidated and their Li-ion transport properties compared to the AB- and diamidoborane borohydrides above. The behaviour of the materials can be rationalised in terms of their composition and hybrid crystal structures.
[LiBH4]2NH3BH3 (1) and Li(BH3NH2BH2NH2BH3) (2) were synthesised following the same procedures reported by Rush et al. and Grochala et al., respectively.16,17 The new lithium AB iodides, LiI(NH3BH3) (3) and LiI(NH3BH3)2 (4) were prepared mechanochemically from stoichiometric quantities of LiI and NH3BH3. A preliminary assessment of results from a series of LiI–NH3BH3 mixtures revealed that different compounds likely formed by varying the molar ratio of the starting materials. Powder X-ray diffraction (PXD; ESI Fig. S14†) revealed that milling of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 LiI:AB mixtures produced two new crystalline phases, nominally LiI(NH3BH3) (3) and LiI(NH3BH3)2 (4). Given the distinctive B–H and N–H stretching (ν) and deformation (δ) modes, Raman spectroscopy is an excellent probe for the study of AB-containing materials. Brief inspection of the spectra strongly suggests that AB is intact within both 3 and 4 (and that B–N–H species exist in 1 and 2 as expected) (ESI†).
:1 and 1:2 LiI:AB mixtures produced two new crystalline phases, nominally LiI(NH3BH3) (3) and LiI(NH3BH3)2 (4). Given the distinctive B–H and N–H stretching (ν) and deformation (δ) modes, Raman spectroscopy is an excellent probe for the study of AB-containing materials. Brief inspection of the spectra strongly suggests that AB is intact within both 3 and 4 (and that B–N–H species exist in 1 and 2 as expected) (ESI†).
The indexing of laboratory PXD patterns was confirmed from synchrotron PXD data. The latter were used for precise structure refinement (ESI†). The new phases 3 and 4 both crystallise in the monoclinic system; the former in space group P21/c (a = 4.39140(1) Å, b = 16.17291(5) Å, c = 7.15685(2) Å, β = 101.52(2)°), the latter in space group C2 (a = 12.40709(4) Å, b = 7.22117(3) Å, c = 4.42025(2) Å, β = 106.21(1)°). The iodine position and good estimates for the B and N positions were found after solving each of the structures with the charge flipping algorithm in Superflip.18 Li positions in 3 and 4 were subsequently tentatively located from difference Fourier maps. The structures of 1–4 are shown in Fig. 1.
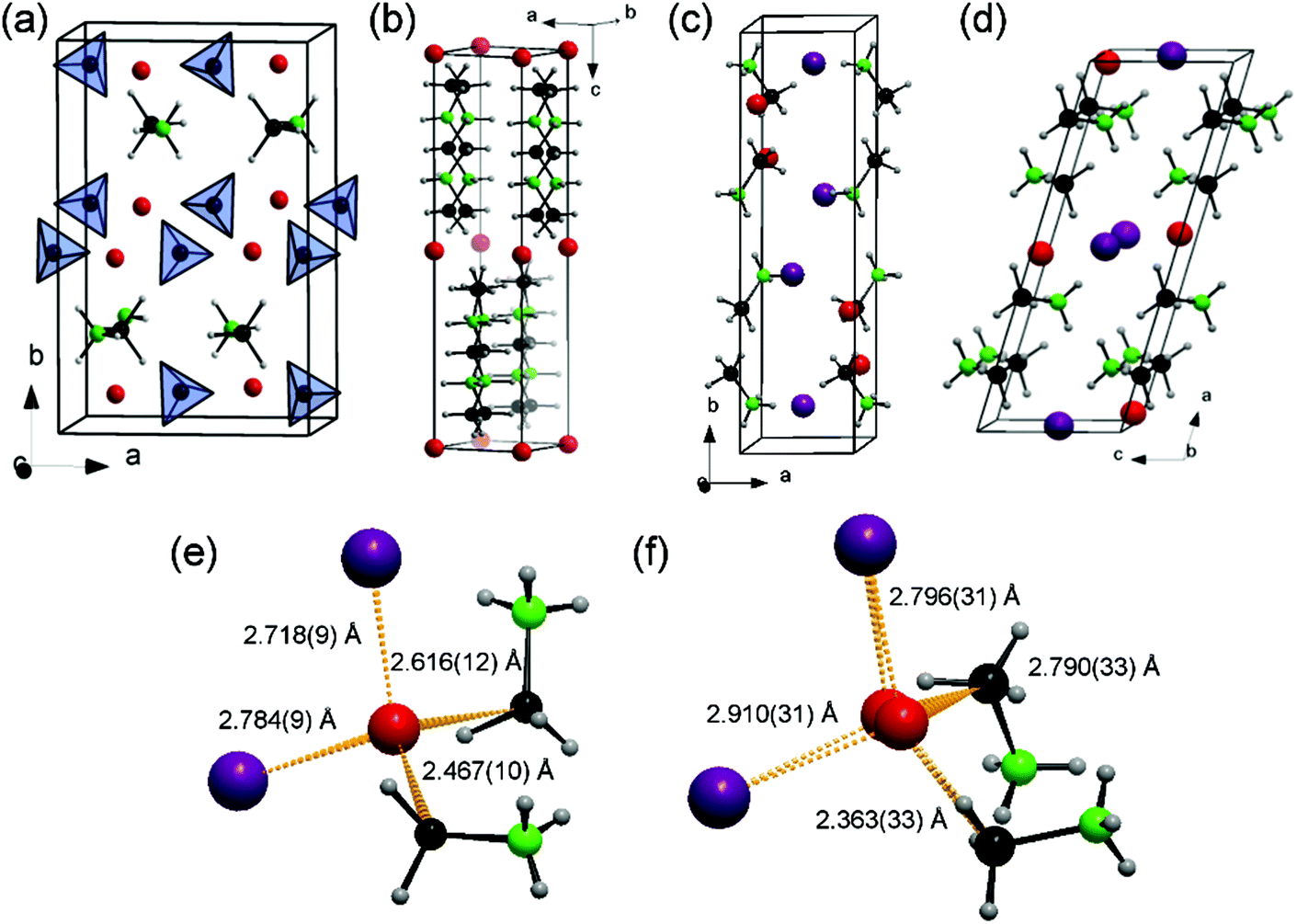 | ||
| Fig. 1 Crystal structures of: (a) [LiBH4]2[NH3BH3] (1); (b) Li(BH3NH2BH2NH2BH3) (2); (c) [LiI][NH3BH3] (3) and (d) [LiI][NH3BH3]2 (4). Detail of the distorted tetrahedral coordination environment of Li+ in (e) [LiI][NH3BH3] (3) and (f) [LiI][NH3BH3]2 (4). Li, I, N, B and H are represented as red, purple, green, black and grey spheres, respectively. | ||
Combined thermogravimetric-differential thermal analysis (TG-DTA) experiments were performed on both new materials 3 and 4 to determine their thermal stability and decomposition behaviour prior to performing AC impedance measurements (Table 1 and ESI†). Both 3 and 4 exhibit sharp endotherms above 100 °C with no concurrent mass loss (with maxima at 119.5 °C and 114.2 °C, respectively) and the two compounds each demonstrate two exothermic decomposition steps thereafter between ca. 120–200 °C (with mass losses of ca. 13 and 16.5 wt%, respectively). The PXD patterns collected for 3 and 4 after the two decomposition steps show the formation of LiI as the only crystalline product (ESI Fig. S19 and S20†), suggesting that both exothermic events originate from the decomposition of AB. This evidence would also imply that the lower temperature endothermic event in the profiles of 3 and 4 likely arises from the decomposition of the compounds into their respective ionic (LiI) and molecular (AB) components (coupled with melting of AB). The thermal stability of 1 and 2 were measured for comparison (Table 1 and ESI†). Both materials also exhibit mass losses between 120–200 °C as has been documented previously.16,17 We note that in both 1 and 2 these decomposition steps are exothermic as seen in 3 and 4 and that in each case the exothermic process is preceded by an endothermic one (at ca. 111 °C and 136 °C for 1 and 2, respectively). We again associate the endotherms with decomposition (for 1) and/or melting. Hydrogen was the predominant gaseous decomposition product but borazine, diborane and ammonia were also evolved.
| Material | ||||||
|---|---|---|---|---|---|---|
| 1 | [LiBH4]2NH3BH316 |
2 | Li[BH3NH2BH2NH2BH3]17 | 3 | 4 | |
| a Not reported. b Amorphous. | ||||||
| Formula mass/g mol−1 | 74.46 | 74.46 | 81.38 | 81.38 | 164.81 | 195.46 |
| 1st endotherm peak T/°C | 97.8 | 136.1 | — | 119.5 | 114.2 | |
| Mass loss onset T/°C | 111.2 | 105 | 136.1 | 140 | 132.3 | 123.5 |
| Mass loss/wt% | 27.4 | 27 | 34.0 | 9 | 11.9 | 16.4 |
| Gases evolved | H2, NH3, B3H6N3, B2H6 | H2, NH3, B3H6N3, B2H6 | H2, B3H6N3, B2H6, NH3 | H2 | H2, B3H6N3, NH3, B2H6 | H2, B3H6N3, NH3, B2H6 |
| Decomposition products (XRD) | LiBH4 | LiBH4 | LiBH4 | LiI | LiI | |
| Decomposition products (Raman) | (NHBH)n | (NH2BH2)n (NHBH)n | (NH2BH2)n (NHBH)n | (NHBH)n | ||
| LiBH4 | LiBH4 | |||||
All conductivity measurements were performed from room temperature to the point of the first endothermic events (the respective melting/decomposition points). Activation energies were extracted from Arrhenius plots for 1 and 2 between 343–403 K and 3 and 4 between 293–343 K, respectively. Above these upper temperature limits further evidence of decomposition was observed in the conductivity data. For 1, for example, where thermal analysis suggested two endotherms prior to mass loss (ESI Fig. S4†), a steep drop in conductivity is observed at ca. 95–100 °C. This is commensurate with decomposition of 1 into AB and LT-LiBH4 (the “low temperature” polymorph) and the subsequent transition of the LT-phase to HT-LiBH4 before AB begins thermal decomposition. The conductivity data for 1–4 are shown in Fig. 2 and Table 2.
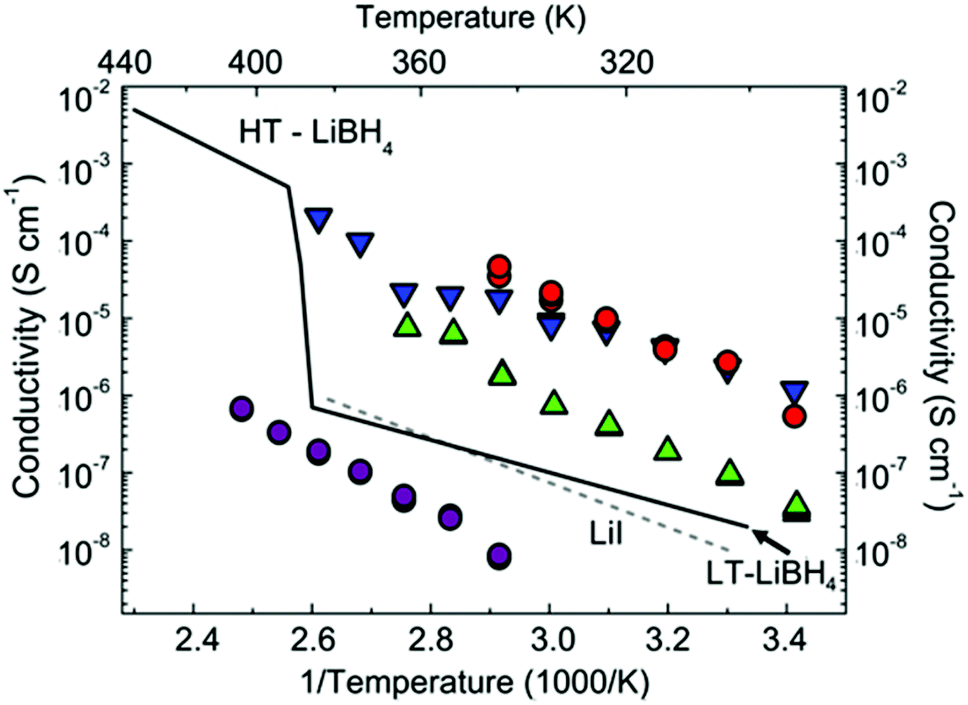 | ||
| Fig. 2 Arrhenius plots of total conductivity against reciprocal temperature (and temperature) for 1–4. The plotted conductivity represents the sum of the grain boundary and bulk contributions. The temperature dependence of the conductivity for LiI7 and LiBH419 is shown for comparison. 1–4 are represented in red, magenta, green and blue respectively. | ||
| Material | σ 343/S cm−1 | E a/eV |
|---|---|---|
| [LiBH4]2[NH3BH3] (1) | 2.83 × 10−5 | 0.71(3) |
| Li[BH3NH2BH2NH2BH3] (2) | 4.11 × 10−9 | 0.86(1) |
| [LiI][NH3BH3] (3) | 1.80 × 10−6 | 0.69(1) |
| [LiI][NH3BH3]2 (4) | 1.79 × 10−5 | 0.48(1) |
Impedance data analysed in the complex plane showed a semicircle at high frequencies and a linear response at low frequencies characteristic of ionic conductivity and ion blocking electrodes. At lower temperatures two semicircles were observed, suggesting intra- and inter-grain transport could be resolved. However, the high frequency semicircle moved beyond the measurement range at higher temperatures and so the data were analysed using an equivalent electrical circuit to extract the values for the total resistivity. 2 has conductivity several orders of magnitude lower than that of the other materials, with a correspondingly higher activation energy for Li+ ion conduction.
The remaining compounds, containing molecular NH3BH3 and either I− or [BH4]− as spherical anions, exhibit superior conductivities with the best performance demonstrated by 1 and 4. In fact, the activation energy for 4 is slightly lower than the values reported for the fast ion conductors HT-LiBH4 (0.53 eV)20 and Li3(NH2)2I (0.58 eV).10 The ionic conductivity for 4 increases by ca. 2 orders of magnitude between 343–383 K and the absolute value is only slightly lower than that observed for iodide-stabilised LiBH4 at these temperatures.
It is well-known that open structures with available interstitial positions are often favourable for ionic conductivity.21 It is perhaps not surprising, therefore that 2 has the lowest Li+ ion conductivity of the materials studied given the relatively bulky anions and the restricted space available for lithium to diffuse. The closest Li–Li distance in 2 is 4.02(1) Å and the average separation between [BH3NH2BH2NH2BH3]− species within anionic layers in the crystal structure of 2 is ∼4.0 Å which is likely to be insufficient to allow the Li+ ions to pass smoothly.17,22 The “pseudo-layered” materials, 1 and 4, exhibit the best ionic conductivity at all temperatures. It is interesting to note that the “ionic section” of the structure of 1 closely resembles the anion and cation arrangement in the fast ion conducting hexagonal HT phase of LiBH4 rather than that in orthorhombic LT-LiBH4, where the conductivity is 103–104 times lower. Hence, integrating AB into the LiBH4 lattice (or alternatively “implanting” LiBH4 within the AB lattice) effectively drives the LT-HT phase transition at reduced temperature much as halide substitution does in LiBH4 itself. One might assume therefore that the Li+ conduction mechanism is also similar and would rely on the cooperative rotational motion of the [BH4]− anions. In compound 1, the BH4− complex anions and NH3BH3 molecules both act effectively as bridging ligands to connect the Li+ ions in three dimensions.23 The closest Li–Li distances are 3.42 Å and 4.05 Å within the LiBH4-type ionic pseudo-layers and 4.30 Å across AB layers. It seems reasonable to envisage anisotropic Li+ ion conductivity in 1 with faster transport within the ionic pseudo-layers (ac plane) than between them (b-direction).
Of the iodides, although 4 contains proportionately less lithium, it has a higher total ionic conductivity across the entire measured temperature range. The segregation of the ionic and molecular sections of the structure in the AB-rich compound 4 are more pronounced than in 3 and similarly to 1, it contains ionic and molecular pseudo-layers. By contrast to 1 however, the molecular layers in 4 are thicker – 2 AB molecules across (along the long a-axis) – and so the structure of 4 is more “AB-like” than 1, with the segregation of the ionic and molecular parts of the structure seemingly facilitating the migration of the Li+ ions in the bc plane in 4. There is no such segregation in 3 and the pathways for Li+ ion conduction would appear to be more restricted and probably limited to one dimension only. Moreover, the structure solution of 4 suggests disorder and hence Li+ vacancies in the lithium sub-lattice, providing a further possible rationale for the enhanced ionic conductivity. Given the relatively weak X-ray scattering of Li+ (in the presence of I−) and the similarity in scattering power of N and B, it is not possible to draw more definitive conclusions regarding the existence of Li+ vacancies or whether the AB molecules in 3 and 4 exhibit dynamic (or positional) disorder. From current evidence, it appears that the AB does not play an active part in a cooperative Li+ ion conductivity mechanism. We intend to clarify this once powder neutron diffraction experiments have been completed. However, the fact that the Uiso values of the B and N atoms become physically unreasonable when the atomic positions are exchanged indicates that the AB molecules may be relatively static. An interesting comparison can nonetheless be made to lithium amide halides such as Li3(NH2)2I (σ ≈1.7 × 10−5 S cm−1 at room temperature). The Li+ ion conductivity in Li3(NH2)2I was proposed to be associated with short Li–Li distances and abundant interstitial sites, rather than being influenced by NH2− reorientation (for which there was no direct evidence).10,24 Compound 1 contains a relatively short Li–Li distance (3.42 Å); within the range of distances observed in Li3(NH2)2I.10,24 However, conversely, compound 4 with comparable conductivity to 1, has no such short Li–Li spacings (with a closest Li–Li distance of ca. 4.3 Å), suggesting that the conductivity mechanisms in the AB borohydride and the AB iodides could indeed be different.
In this context, it is also useful to compare the conductivity of 3 and 4 with LiI itself. Rock salt-structured LiI shows only modest ionic conductivity at room temperature.25 This is despite the fact that the anion has the highest polarizability among the halides.26 The observed ionic conductivity of LiI is predominantly extrinsic below 180 °C,25 whereas above 180 °C, its intrinsic conducting behaviour is mediated by Schottky defects. This can be dramatically enhanced in the monohydrate (LiI·H2O; with an anti-perovskite structure), where 2/3 of the octahedral Li+ sites are vacant (or equally the conductivity can be improved by aliovalent doping of Ca2+ in LiI).27 By loose analogy to the hydrate, inserting LiI into a lattice of AB should enlarge the space for Li migration (providing new and available interstitial sites) and enhance ionic conductivity. This would especially appear to be the case in 4 where the activation energy for Li+ transport is notably lower than in any of the other materials.
Experimental
Full details are provided in the ESI.† [LiBH4]2NH3BH3 (1) and Li(BH3NH2BH2NH2BH3) (2) were synthesised following previously reported procedures.16,17 The LiI–NH3BH3 materials (3 and 4) were prepared by ball milling stoichiometric ratios of LiI and NH3BH3 in hardened steel jars (50 ml) with 10 hardened steel balls of 10 mm diameter using a Retsch PM100 ball mill. The mixture was milled for 2 h at 250 rpm (400:1, ball-to-powder-ratio by mass) to yield white powders.
Powder X-ray diffraction (PXD) data were obtained at room temperature with a Bruker D8 Advance (θ–2θ) diffractometer, using Cu Kα1 radiation and a step size of 0.017° 2θ over 5–95° 2θ for ca. 12 h. Jana2006 was used for the initial Le Bail fitting and space group assignments of the new compounds 3 and 4.28 The structures of 3 and 4 were then solved using Superflip within Jana. For 4, Li positions were tentatively identified from difference Fourier maps. High Resolution synchrotron PXD experiments were performed using beamline I11 (λ = 0.826281(10) Å) and the structures refined against the synchrotron data using the Rietveld method (with GSAS/EXPGUI software).29,30
Raman spectra were measured at room temperature using a Horiba Jobin Yvon Raman microscope with a green laser (λ = 532 nm) and employing a hole aperture of 50 μm, 100 g mm−1 grating and a synapse CCD detector. Sample morphology and composition were studied using scanning electron microscopy (SEM) (XL 30 ESEM, Philips, 25 kV accelerating voltage with an Oxford Instruments X-act spectrometer for EDX analysis). Thermogravimetric-differential thermal analysis-evolved gas mass spectrometry (TG-DTA-MS; NETZSCH STA 409PC and HIDEN HPR20 mass spectrometer) was performed from room temperature to 473 K at 5 K min−1 under an Ar flow. MS signals were monitored with time and temperature.
Electrochemical impedance measurements (Solartron 1260 impedance analyser) were performed on cylindrical pellets on heating, allowing the temperature to equilibrate for ≥1 h before collecting data. Data were analysed using equivalent circuit analysis as implemented in the ZView2 software package.
Conclusions
In summary, ammonia borane halides have been synthesised for the first time and the 2 new iodides can be classified with [LiBH4]2NH3BH3 into a broader family of hybrid compounds, [LiX]m(AB)n (X = I−, BH4−; m, n = integer). The materials are stable above 100 °C and decompose to the binary halides (borohydride) with the evolution of hydrogen as the main gaseous product. The Li-ion conductivity of these hybrid hydrides has been determined and exceeds that of the binary respective ionic components by orders of magnitude. The conduction pathways can be tailored through the partition of ionic and molecular sections in these hybrid materials. In principle, Li-ion transport in this class of materials could be further optimised by substitutions in the ionic sub-structure. Moreover, the control of static and dynamic disorder in the cationic, anionic and molecular sub-lattices should prove pivotal in the design of new Li+ ion conductors and ultimately potential electrolytes. Indeed, a vast range of new hybrid materials should be accessible with transport properties and stabilities that can be manipulated by the judicious selection of all the component species; cationic, anionic and molecular.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Universities of Glasgow and Strathclyde for supporting this work and Mr Nicolás Flores González for assistance with MS data. The synchrotron radiation experiments were performed at the I11 instrument at the Diamond Light Source, Oxfordshire (beamtime award EE13155).Notes and references
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS PubMed.
- T. K. Mandal and D. H. Gregory, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2009, 105, 21 RSC.
- T. K. Mandal and D. H. Gregory, Proc. IMechE, Part C: J. Mech. Eng. Sci., 2010, 224(C3), 539 CrossRef.
- H. Reardon, J. Hanlon, R. W. Hughes, A. Godula-Jopek, T. K. Mandal and D. H. Gregory, Energy Environ. Sci., 2012, 5, 5951 RSC.
- M. Matsuo and S. Orimo, Adv. Energy Mater., 2011, 1, 161–172 CrossRef CAS.
- P. E. Jongh, D. Blanchard, M. Matsuo, T. J. Udovic and S. Orimo, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 1 CrossRef.
- H. Maekawa, M. Matsuo, H. Takamura, M. Ando, Y. Noda, T. Karahashi and S. Orimo, J. Am. Chem. Soc., 2009, 131, 894 CrossRef CAS PubMed.
- I. Cascallana-Matías, D. A. Keen, E. J. Cussen and D. H. Gregory, Chem. Mater., 2015, 27, 7780 CrossRef.
- M. Matsuo, A. Remhof, P. Martelli, R. Caputo, M. Ernst, Y. Miura, T. Sato, H. Oguchi, H. Maekawa, H. Takamura, A. Borgschulte, A. Züttel and S. Orimo, J. Am. Chem. Soc., 2009, 131, 16389 CrossRef CAS PubMed.
- M. Matsuo, T. Sato, Y. Miura, H. Oguchi, Y. Zhou, H. Maekawa, H. Takamura and S. Orimo, Chem. Mater., 2010, 22, 2702 CrossRef CAS.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28 CrossRef CAS PubMed.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 25, 2613 RSC.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279 RSC.
- A. Staubitz, A. P. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079 CrossRef CAS PubMed.
- Z. Xiong, C. K. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson and P. P. Edwards, Nat. Mater., 2008, 7, 138 CrossRef CAS PubMed.
- H. Wu, W. Zhou, F. E. Pinkerton, M. S. Meyer, G. Srinivas, T. Yildirim, T. J. Udovic and J. J. Rush, J. Mater. Chem., 2010, 20, 6550 RSC.
- K. J. Fijalkowski, T. Jaron, P. J. Leszczynski, E. Magos-Palasyuk, T. Palasyuk, M. K. Cyranski and W. Grochala, Phys. Chem. Chem. Phys., 2014, 16, 23340 RSC.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- D. Sveinbjornsson, J. S. G. Myrdal, D. Blanchard, J. J. Bentzen, T. Hirata, M. B. Mogensen, P. Norby, S. I. Orimo and T. Vegge, J. Phys. Chem. C, 2013, 117, 3249 CrossRef CAS.
- W. Li, G. T. Wu, Z. T. Xiong, Y. P. Feng and P. Chen, Phys. Chem. Chem. Phys., 2012, 14, 1596 RSC.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363 CrossRef CAS.
- R. Owarzany, K. J. Fijalkowski, T. Jaroń, P. J. Leszczyński, Ł. Dobrzycki, M. K. Cyrański and W. Grochala, Inorg. Chem., 2015, 55, 37 CrossRef PubMed.
- M. Paskevicius, L. H. Jepsen, P. Schouwink, R. Černý, D. B. Ravnsbæk, Y. Filinchuk, M. Dornheim, F. Besenbacher and T. R. Jensen, Chem. Soc. Rev., 2017, 46, 1565 RSC.
- A. V. Skripov, R. V. Skoryunov, A. V. Soloninin, O. A. Babanova, M. Matsuo and S. Orimo, J. Phys. Chem. C, 2015, 119, 13459 CrossRef CAS.
- F. W. Poulsen, Solid State Ionics, 1981, 2, 53 CrossRef CAS.
- A. R. West, Basic Solid State Chemistry, John Wiley & Sons Ltd, Chichester, 1988 Search PubMed.
- G. Eichinger, Solid State Batteries, chapter: conductivity of modified lithium iodide samples, Springer Netherlands, Dordrecht, 1985 Search PubMed.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345 CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR, 1994, 86.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qi00099b |
| ‡ Present address: Structure and Dynamics of Energy Materials, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany. |
| § Present address: Dept of Chemical & Biological Engineering, University of Sheffield, Mappin Street, Sheffield S1 3JD, UK. |
| This journal is © the Partner Organisations 2019 |