
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-containing hybrid compounds based on extremely rare [V2Mo6O26]6– POM as water oxidation catalysts†
Halyna I.
Buvailo
a,
Valeriya G.
Makhankova
 *a,
Vladimir N.
Kokozay
a,
Irina V.
Omelchenko
b,
Svitlana V.
Shishkina
b,
Julia
Jezierska
c,
Mariia V.
Pavliuk
d and
Sergii I.
Shylin
*d
*a,
Vladimir N.
Kokozay
a,
Irina V.
Omelchenko
b,
Svitlana V.
Shishkina
b,
Julia
Jezierska
c,
Mariia V.
Pavliuk
d and
Sergii I.
Shylin
*d
aDepartment of Chemistry, Taras Shevchenko National University of Kyiv, Volodymyrska 64/13, 01601 Kyiv, Ukraine. E-mail: leram@univ.kiev.ua
bInstitute for Single Crystals, National Academy of Sciences of Ukraine, Nauky Ave 60, 61001 Kharkiv, Ukraine
cFaculty of Chemistry, University of Wroclaw, F. Joliot-Curie 14, 50-383 Wroclaw, Poland
dDepartment of Chemistry – Ångström Laboratory, Uppsala University, P.O. Box 523, 75120 Uppsala, Sweden. E-mail: sergii.shylin@kemi.uu.se
First published on 29th May 2019
Abstract
Herein, we report two approaches to the synthesis of heterometallic complexes (NH4)2n(H2en)n{[Cu(en)2][α-V2Mo6O26]}·4nH2O (1), (NH4)2{[Cu(dien)(H2O)]2[α-V2Mo6O26]}·5H2O (2) and (NH4)2{[Cu(dien)(H2O)]2[α-V2Mo6O26]}·8H2O (3) that have been employed in homogeneous photochemical oxidation of water to dioxygen. In these hybrid metalorganic–inorganic compounds, copper-containing complex fragments are covalently bound to the rare vanadium-disubstituted α-octamolybdate cluster. They exhibit variable catalytic activity controlled by the local coordination environment of copper reaching a notably high turnover frequency of 0.24 s−1 for 3 in combination with a relatively low water oxidation overpotential. The complexes have been also used as precursors for the preparation of mixed oxide phases that have proven to be active heterogeneous water oxidation catalysts.
Introduction
The development of technologies for the production of chemical fuels or useful compounds utilizing renewable energy and raw materials relies on a sustainable supply of protons and electrons to form the desired products.1 Climate change driven by the increasing fossil-based energy consumption provides a strong motivation for research into the development of carbon-neutral and carbon-negative energy systems. In this regard, various approaches harnessing sunlight as a practically infinite energy source are becoming realistic and appealing solutions for achieving the goals for a sustainable society.2 Existing solar photovoltaic technologies can convert light energy from the sun into electricity. However, the sunlight is intermittent and unevenly spread over the entire surface of the planet. It requires methods for storage of solar energy in chemical bonds in the form of dense and transportable green fuels. Artificial photosynthesis using water as an abundant source of electrons which can be recombined at the point of use in a fuel cell is becoming one avenue to solve the energy storage problem.3 To date, the development of water oxidation catalysts (WOCs) remains a challenging task to attain efficient solar energy conversion since oxidation of water to dioxygen is a main bottleneck for the overall water splitting.4 It is a complicated process involving four-electron transfer coupled with four-proton transfer. Hence, the WOC must possess rich redox chemistry enabling an array of multielectron oxidations to evolve dioxygen from two water molecules.5For the past few decades, various heterogeneous WOCs considered as suitable for practical applications and their homogeneous analogues allowing to study the mechanism of catalysis have been designed. Ruthenium and iridium oxides are well-known efficient water oxidation catalysts.6 They have found the application in commercial proton-exchange membrane electrolyzers for large-scale hydrogen production from renewables.7 Among homogeneous WOCs, molecular complexes of ruthenium and iridium have also proven to be the most active and stable for oxygen evolution.8 However, a low natural abundance and harm to the environment limit the ability of WOCs based on these two noble metals to meet growing global energy demands. In this regard, there is considerable interest in the use of cheap first-row transition metal compounds for water oxidation.9 A handful of manganese,10 iron,11 cobalt,12 nickel,13 and copper complexes have been investigated with respect to their catalytic activity in water oxidation. Among them, copper-containing WOCs are most newly reported and least studied. Mayer et al. were the first to demonstrate the potential of copper complexes in electrochemical water oxidation employing Cu(bpy)(OH)2 (bpy = 2,2′-bipyridine) as a catalyst under alkaline conditions.14 Later studies have showed that other molecular copper compounds could catalyze electrochemical water oxidation, including polypeptide complexes,15 compounds with modified 2,2′-bipyridine and multidentate polypyridine ligands,16 pyridine–alkoxide complexes,17 and amidate complexes.18 However, the eventual goal is to develop the efficient solar light-driven WOCs and to avoid the thermal and/or electrochemical oxidation. Only a few copper-based catalysts active in photochemical water oxidation have been reported to date: a polypyridine complex [Cu(F3TPA)]2+ (F3TPA = tris(2-fluoro-6-pyridylmethyl)amine),19 a macrocyclic square-planar tetrasulfonatophthalocyanine complex,20 and all-inorganic compound [Cu5(OH)4(H2O)2(SiW9O33)2]10– based on polyoxometalate (POM).21 The catalytic activity of the latter can be attributed to its ability to undergo reversible and stepwise multielectron transfer without changing the cluster structure. Moreover, POMs are intensively studied class of inorganic materials that can be considered as soluble ultrasmall oxide nanoparticles.22 They hold great promise for a variety of applications in catalysis,23 medicine, magnetism and photochromism.24 Featuring advantages of homogeneous and heterogeneous catalysts, POMs have been explored as efficient WOCs.25 In recent years, various approaches aimed at decorating the anionic POM clusters with complex cations have been developed.26 In this way, one can tune catalytic, optical, electrochemical and magnetic properties of the material.27 Previously, we have successfully isolated 3d transition metal-containing polyoxovanadate and polyoxomolybdate hybrid compounds using one-pot direct synthesis approach.28,29 The complexes of manganese and cobalt decorated with decavanadate clusters have proven to be efficient homogeneous WOCs.29 In order to modify the catalytic performance of the hybrid WOCs, we have obtain three heterometallic compounds constructed from copper-polyamine fragments coordinated by the rare [V2Mo6O26]6– cluster anions. Their activity has been evaluated in the model photo-oxidant system using [Ru(bpy)3]2+ dye as photosensitizer and persulfate as sacrificial electron acceptor (Fig. S1†). The compounds exhibit different coordination manner of copper by the polyamine ligands and water molecules providing insights into the water oxidation mechanism. As there is considerable interest in the use of heterogeneous WOCs for developing solar energy conversion devices, the mixed–oxide phases obtained after thermal degradation of the complexes have been also examined in photochemical water oxidation appropriate.
Results and discussion
Synthesis and characterization of hybrid compounds
Hybrid metalorganic–inorganic compounds based on POMs are usually synthesized starting from solutions of corresponding building blocks (e.g., metal complexes and POM clusters) or by hydrothermal techniques. Both methods often give rise to formation of a mixture of products, which require further purifications. Following our previous studies on the direct synthesis approach,28–30 we have obtained three hybrid compounds (NH4)2n(H2en)n{[Cu(en)2][α-V2Mo6O26]}·4nH2O (1) (en = ethylenediamine), (NH4)2{[Cu(dien)(H2O)]2[α-V2Mo6O26]}·5H2O (2) (dien = diethylenetriamine) and (NH4)2{[Cu(dien)(H2O)]2[α-V2Mo6O26]}·8H2O (3) starting from copper metal, (NH4)6Mo7O24·4H2O, V2O5 and corresponding polyamine from aqueous solution. This kind of synthesis occurs under mild and anion-deficient conditions promoting selective formation of POMs that can be easily isolated from the mother liquid (see Experimental section for details). When en is used as a ligand, the target product 1 can be obtained in the course of the copper dissolution accompanied by air oxidation only. Using the wet chemistry method (i.e., starting from soluble copper salts instead of copper metal), we were not able to isolate a crystalline product. When dien is used as a ligand, the oxidative dissolution of copper leads to formation of green crystals of 2. After crystalline product 2 has been filtered off, violet crystals of 3 start to precipitate from the mother liquid. The purity of 2 and 3 obtained from the same reaction mixture has been confirmed by PXRD analysis; the positions of the experimental peaks are in good agreement with the simulated peak profiles calculated from the single-crystal XRD data (Fig. S2†). In contrast to 1, products 2 and 3 could be isolated using both direct synthesis and wet chemistry method. However, the higher yields of the final compounds were achieved using Cu0 as a starting copper source.Crystal structure
Single-crystal X-ray structure analysis reveals that three obtained compounds are based on rare vanadomolybdate cluster [V2Mo6O26]6–. It has crystal structure similar to that for trivial α-octamolybdate anion [Mo8O26]4–.31 However, the V-substituted cluster consists of two {VO4} units capping opposite faces of a ring constructed from six edge-sharing {MoO6} octahedra leading to overall D3d symmetry. At first glance, the ring of octahedra caped with two tetrahedra looks like well-known phosphomolybdate Strandberg anion32 but having six {MO6} polyhedra in the ring instead of five. More symmetrical anion is formed due to a larger covalent radius of V heteroatom as compared to P. The similar POMs with even larger As heteroatom have been also reported.33 Both [X2Mo6O26]6– (X = V, As) anions are extremely rare in contrast to their phosphorous analogue. Though the vanadomolybdate cluster was first reported in 1979,34 to our knowledge, there are just a few examples of hybrid compounds based on this V-substituted POM reported to date.35 Importantly, in the anion each {VO4} tetrahedron shares its three O atoms with the polyoxomolybdate ring, while the fourth O atom is a potential coordination or protonation site of the cluster. The Mo–O distances for three types of O atoms are notably different (Table S1†). For terminal O atoms, Mo–O distances range from 1.675(3) to 1.723(4) Å, indicating double Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. However, Mo–μ2-O and Mo–μ3-O distances are 1.887(3)–1.930(3) Å and 2.270(3)–2.368(5) Å, respectively, corresponding to single Mo–O bonds. Double bond lengths of V–O (terminal) are from 1.624(3) to 1.660(6) Å while V–μ3-O single bonds are from 1.726(5) to 1.766(3) Å. These structural features of the anions in 1–3 are typical for the octamolybdate cluster, whose capping tetrahedra contain heteroatoms.35
O bonds. However, Mo–μ2-O and Mo–μ3-O distances are 1.887(3)–1.930(3) Å and 2.270(3)–2.368(5) Å, respectively, corresponding to single Mo–O bonds. Double bond lengths of V–O (terminal) are from 1.624(3) to 1.660(6) Å while V–μ3-O single bonds are from 1.726(5) to 1.766(3) Å. These structural features of the anions in 1–3 are typical for the octamolybdate cluster, whose capping tetrahedra contain heteroatoms.35
In 1, the CuII coordination sphere consists of four N atoms from two en molecules in the equatorial plane and two O atoms from two POMs in the axial positions of a distorted coordination octahedron (Fig. 1, Table S1†). The {Cu(en)2} unit is linked to the cluster through terminal oxygen atom from {MoO6} fragment with Mo–O(Cu) distance of 1.723(3) Å. The anion and copper moieties are arranged alternately forming an infinite 1D chain parallel to the [011] crystallographic direction with the chain pitch of 13.769 Å. It is additionally stabilized by N–H⋯O contacts between H atoms of coordinated en and O atoms of POM anion (Fig. S3, Table S2†). The voids between the chains are filled with ammonium and non-coordinated H2en2+ cations as well as water molecules, linked to the chain and to each other by the series of hydrogen bonds.
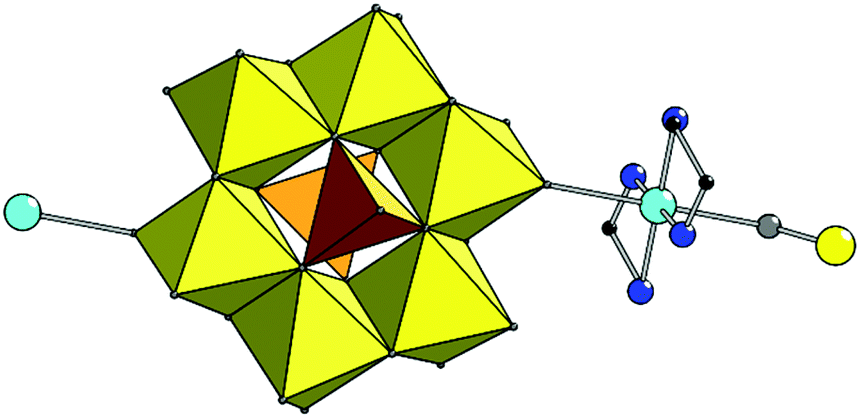 | ||
| Fig. 1 Combined polyhedral and ball-and-stick representation of the 1. H atoms are omitted for clarity. Colour scheme: Mo yellow, V orange; Cu cyan; N blue; C black; O grey (this colour scheme is used throughout the manuscript). | ||
Both structures 2 and 3 have complex anions {[Cu(dien)(H2O)]2[V2Mo6O26]}2– of exactly the same composition, formed by [V2Mo6O26]6– linked to two {[Cu(dien)(H2O)]} complex cations. Copper site in {[Cu(dien)(H2O)]} fragment reveals (4 + 1) coordination type forming {[CuON3]O} coordination polyhedron with three nitrogen donors of one dien ligand and one O atom of water in equatorial plane and with other O atom at a larger distance from Cu in the axial position (Table S1,†Fig. 2).
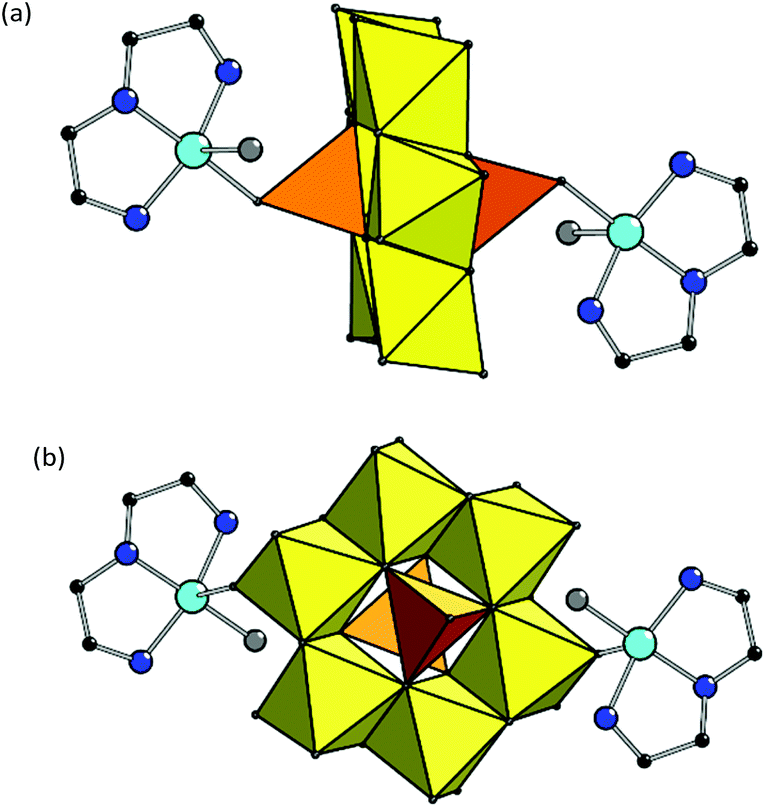 | ||
| Fig. 2 Combined polyhedral and ball-and-stick representation of the complex anions {[Cu(dien)(H2O)]2[V2Mo6O26]}2– in 2 (a) and 3 (b). H atoms are omitted for clarity. | ||
The compounds differ in a manner of binding of copper fragments to POM. In 2, the coordinated oxygen in equatorial plane belongs to the [VO4] polyhedra of the POM fragment (Cu–O(V) bond is 1.983(6) Å), while the axial position – to the water molecule (Cu–OH2 distance is 2.380 Å). In 3, another coordination manner is observed: O atom in equatorial plane is from the water molecule (Cu–OH2 distance is 1.966 Å) while the other one is the terminal O atom of [MoO6] polyhedra of the POM anion (Cu–O(Mo) is 2.416 Å). Such a substantial difference in the values of Cu–OH2 bonds in 2 and 3 is due to the various positions of coordinated H2O in the copper complexes.36 The value of angular structural index parameter τ is 0.26 in 2 and 0.22 in 3, therefore, the {[CuON3]O} coordination polyhedra have square-based pyramidal geometry with trigonal bipyramidal distortion.37
In 2, the complex fragments are connected in integrated 3D structure via multiple hydrogen bonds involving ammonia cations and crystallization water molecules (Fig. S4†). In 3, the second axial possible coordination site at Cu centre is occupied by the terminal O atom of the [MoO6] polyhedron of neighboring POM anion with significantly elongated Cu–O distance of 2.889 Å which can be interpreted as supramolecular interaction. The copper fragments are connected with POM forming 1D chain along [010] direction (Fig. 3). Moreover, the chain is additionally stabilized by H-bonds by means of H atoms of coordinated water molecules together with terminal O atoms of vanadate fragments (Table S2†); the chain pitch is 9.195 Å. The chains are linked via hydrogen bonds involving ammonia cations and water molecules located in voids between the chains.
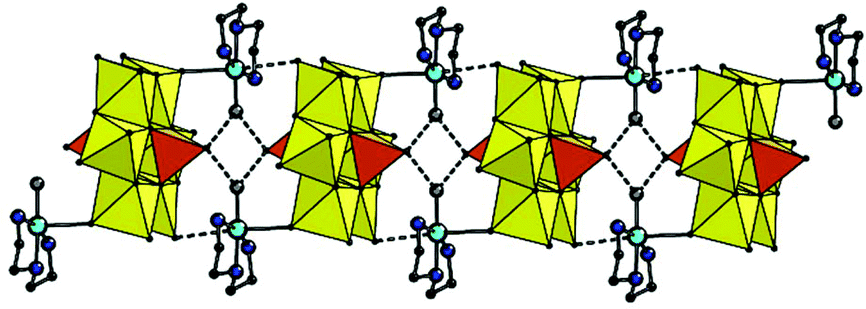 | ||
| Fig. 3 Combined polyhedral and ball-and-stick representation of the supramolecular 1D chain in the structure of 3. The H-bonds and intermolecular Cu–Ot(Mo) contacts are shown as dashed lines. H atoms, non-coordinated water molecules and ammonium cations are omitted for clarity. | ||
FTIR and UV-vis spectroscopy features
The FTIR spectra of the compounds 1–3 reflect the formation of the V-substituted octamolybdate cluster (Fig. 4).35a The intensive bands between 1000 and 500 cm−1 can be assigned to M–O and M–O–M (M = Mo, V) stretching vibrations of the POM. The bands around 1610 cm−1 and 1420 cm−1 are characteristic of in-plane bending δ(HOH) of water molecules and δ(HNH) of coordinated en/dien ligands, respectively. The strong absorption for 1 at 1045 cm−1 can be assigned to the C–N modes of the H2en2+ cations located in the interchain space.28a The characteristic bands in the region of 3340–3180 cm−1 are attributed to the N–H vibrations of the amines, while intensive broad bands in the region of 3650–3100 cm−1 are associated with the stretching vibrations ν(O–H) of the water molecules (Fig. S5†). | ||
| Fig. 4 FTIR spectra of 1–3. | ||
The intensive bands in the UV-vis reflectance spectra of the solid state compounds 1–3 observed at ∼320 nm can be attributed to the O → M charge transfer characteristic of POMs (Fig. S6a†).35b Broad bands in the visible region are typical for the d–d transition in divalent copper complexes.38 The difference between the position of the d–d band for 2 (λmax = 630 nm) and 3 (λmax = 560 nm) may originate from the different coordination polyhedra. The elongated sixth bond in 3 leads to the remarkable blue shift of the d–d band that is reflected in variation of color: the crystals of 2 are green while 3 are violet. Moreover, the shift can be also assigned to dissimilar origin of O donor at equatorial positions of Cu complexes in 2 and 3.
Interestingly, two green solutions were obtained upon dissolving of the compounds in water. Their UV-vis absorption spectra appeared to be similar (Fig. S6b†). Although 3 can be regarded as 1D coordination polymer, it is soluble in water – as well as discrete complex 2 – assuming dissociation of the chains. Indeed, one of the Cu–Ot(Mo) contacts in 3 is significantly elongated and arguably breaks upon solvation. In the ESI-TOF mass spectra of the aqueous solutions of 2 and 3 the same complex anion with m/z = 1464 can be found attributed to {[Cu(dien)(H2O)]2[α-V2Mo6O26] + H}−, based on its isotope pattern and molecular weight (Fig. S7†).
EPR spectroscopy
The paramagnetic species formed in the reaction mixture solutions containing dien (see Experimental section), obtained via oxidative dissolution of zero-valent copper (Method 1) and wet method using Cu(II) salts (Method 2), were identified using EPR spectroscopy. The spectra recorded at 77 K exhibit distinctly resolved copper hyperfine splitting signals in the g∥ region (Fig. 5). The spin-Hamiltonian parameters determined by the simulation of the experimental spectra (Table 1) are in agreement with the values reported previously for Cu(II) complexes with four N atoms in an equatorial plane.39a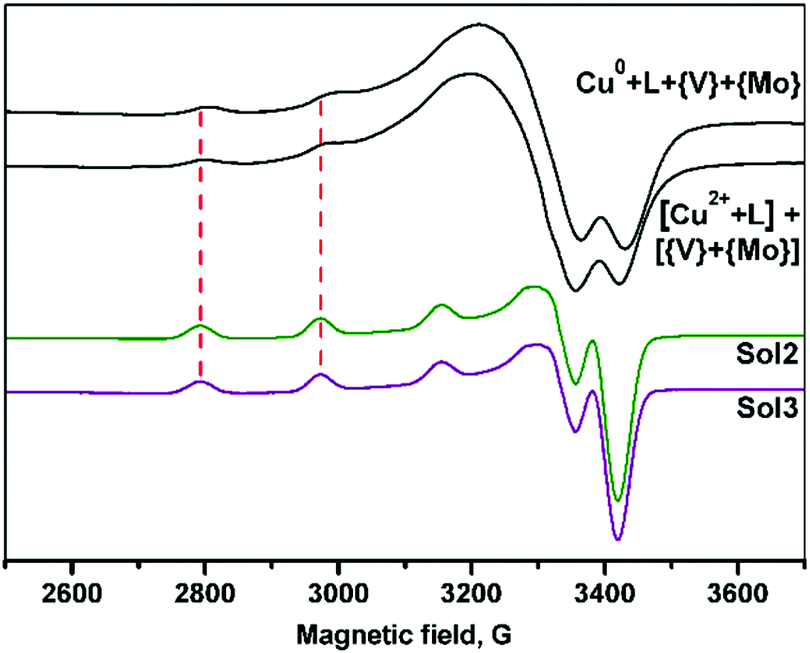 | ||
| Fig. 5 EPR spectra of the frozen aqueous solutions (77 K) of filtrates obtained by Method 1 (starting from Cu0) and Method 2 (the use of Cu2+ salt) and dmso solutions of 2 and 3 (Sol2 and Sol3, respectively). | ||
| Solution | g ∥ | A ∥ 104 cm−1 | Central atom and its equatorial plane |
|---|---|---|---|
| Cu0–dien–V2O5–(NH4)6Mo7O24–H2O (Method 1) | 2.213 | 190 | CuN4 |
| [CuSO4 + dien + H2O] + [V2O5–(NH4)6Mo7O24 + H2O] (Method 2) | 2.210 | 190 | CuN4 |
| Sol2 (DMSO) | 2.234 | 181 | CuN3O |
| Sol3 (DMSO) | 2.234 | 181 | CuN3O |
Slightly acidic media of the reaction mixtures (see experimental part) can promote the protonation of one NH2-group of dien and therefore formation of [CuL2] species39a instead of typical for this ligand [CuL] complexes.39b Thus, despite different reaction pathways, similar copper complexes are present in the both solutions at the final steps of the reaction. In contrast to the previously reported data,28b the reduction of VV to VIV by zero-valent metal was not observed.
The EPR spectra of the 2–3 solutions in dimethyl sulfoxide (dmso) indicated an increase of g∥ and decrease of A∥ values as compared to that observed for Cu(II) complexes formed in the reaction mixtures. It confirms the changes from N4 to N3O donor sets in xy Cu(II) plane.39b The results are in accordance with the structural data indicating that copper fragments remain intact in solutions.
It should be noted that the spin-Hamiltonian parameters of spectra for water and dmso solutions are the same, however, the hyperfine structure is better resolved in dmso.
Thermal properties
Thermal behavior of 1–3 studied by thermogravimetry (TG) and differential thermal analysis (DTA) is presented in Fig. S8.† Compounds 1–3 follow the expected way of decomposition. The initial weight loss occurs in 90–230 °C range and can be attributed to release of the water and ammonia molecules. These processes are characterized by endothermic peaks on the DTA curves. They are followed by the weight loss in the region around 400 °C that is accompanied by the exothermic effect associated with the elimination of the coordinated polyamine ligands and formation of oxide phases. A narrow endothermic peak for 1–3 at ∼600 °C is associated with melting. The detailed description of the thermal degradation can be found in Table S3.†As can be inferred from the XRD patterns (Fig. 6) obtained after the thermal degradation of 2 and 3 in air, their solid products 2-ox and 3-ox contain similar crystalline phases. There is not only MoO3 (orthorhombic) and CuO phases but also Cu–V bronze, which can be tentatively assigned to CuxV2O5 composition40 with lattice parameters a = 28.360, b = 3.628, c = 9.441, β = 98.810. Partial reduction of V(V) most likely occurred due to the presence of volatile organic molecules. In 3-ox, the higher relative content of the bronze phase comparing to 2-ox can influence their properties.
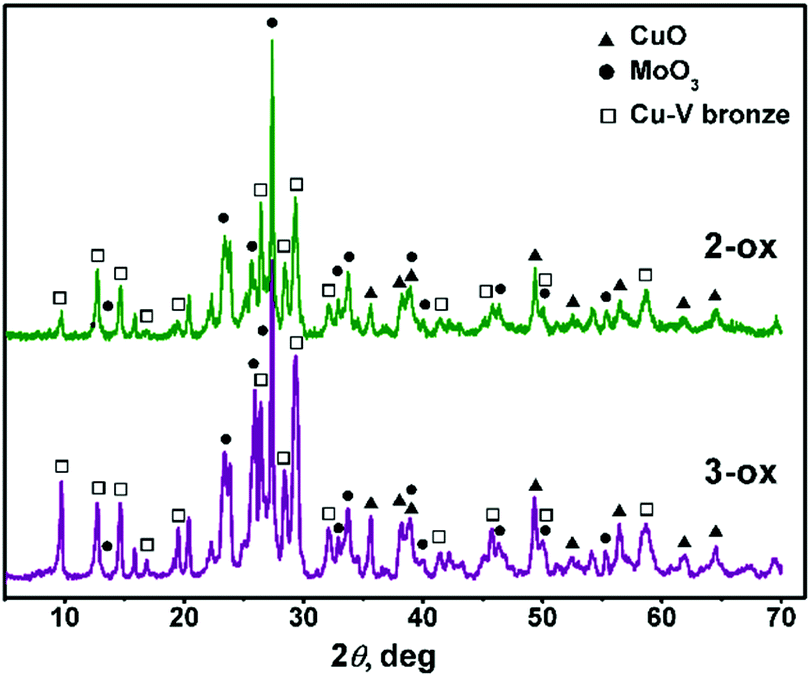 | ||
| Fig. 6 Powder XRD patterns of 2-ox and 3-ox. | ||
Homo- and heterogeneous light-driven water oxidation studies
The catalytic activity of the complexes in photochemical water oxidation was evaluated at pH 8.0. Two compounds 2 and 3 were found to evolve oxygen in the system utilizing [Ru(bpy)3]2+ as a light harvester and persulfate as a sacrificial electron acceptor, while 1 was inactive (Fig. 7). This observation is in harmony with the generally accepted mechanistic pathway for the catalytic water oxidation.15a,41 The initial one-electron oxidation of the catalyst leads to the formation of a CuIII–OH species followed by the subsequent oxidation to CuIVO species. The high-valent metal serves as an attractive substrate for the nucleophilic attack of a water molecule to complete the coordination surrounding of Cu. These aqua-oxo species can easily give rise to the intramolecular O–O bond. The assumed mechanism is feasible for 2 and 3, whose Cu centres are pentacoordinated, and one of positions is occupied by water. In case of 1, the Cu ion is hexacoordinated and cannot yield a key aqua-oxo complex.
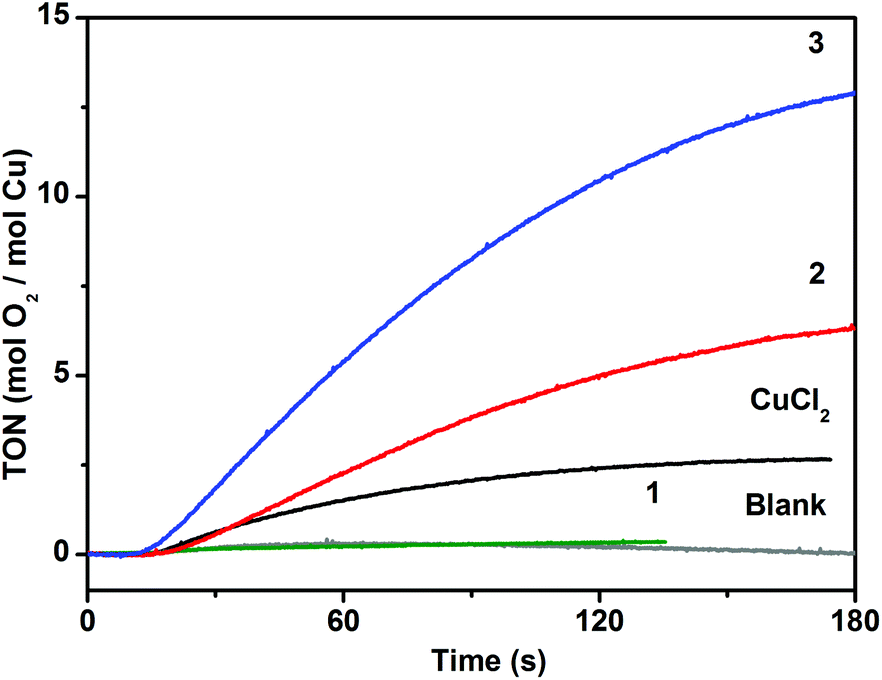 | ||
| Fig. 7 Oxygen evolution traces for the homogeneous catalytic systems consisting of 1, 2, 3 (4 μM) or CuCl2·2H2O (8 μM), [Ru(bpy)3](ClO4)2 (0.1 mM), and Na2S2O8 (2 mM) at pH 8.0 driven by blue light (3 W LEDs, λ = 450 nm). The blank trace (grey line) was obtained by illuminating the solution in the absence of catalyst. | ||
Despite the almost identical composition of 2 and 3, their activity differs almost twofold (values of TON = 7 and 13 and TOF = 0.13 s−1 and 0.24 s−1 for 2 and 3, respectively) that corroborates further the suggested mechanism. The coordinated water molecule has different positions in 2 and 3 in respect to the vacant coordination site. The water molecule and the vacant site are in trans-arrangement in 2 and in cis-arrangement in 3. In the case of 3, formation of the intramolecular O–O bond following addition of a second water molecule is sterically possible, while in 2 this would require structural changes around the Cu ion to facilitate formation of such intermediate. Lloret Fillol et al. have shown that the presence of cis labile sites in molecular Fe-based WOCs is the key structural feature for allowing oxygen evolution.11c They have suggested that formation of the O–O bond takes place via attack of a water molecule assisted by a hydrogen-bond interaction with a hydroxo ligand in a cis-arrangement to a place of the attack. We assume that both, intra- and intermolecular O–O bond formation scenarios are possible. However, taking into account that the trans analogue 2 is catalytically active, we favour the latter mechanism over the intramolecular O–O coupling.
DLS experiments performed within 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) min after the oxygen evolution assay show no indication of nanoparticle formation, which confirms the molecular integrity of the catalysts 2 and 3 (Fig. S9†). We have performed control photochemical water oxidation experiments under nearly identical conditions, but using copper(II) chloride instead of POMs as a WOC. When 8 μM CuCl2·2H2O is used, water oxidation is observed with TON < 3. Albeit there is a limited number of reported studies devoted to investigation of photochemical water oxidation using homogeneous Cu complexes,19–21 to the best of our knowledge, the compounds 2 and 3 exhibit relatively high catalytic efficiency (Table 2). Specifically, their TOF values are three to five times higher compared to the pentacopper tungstosilicate analogue.21
min after the oxygen evolution assay show no indication of nanoparticle formation, which confirms the molecular integrity of the catalysts 2 and 3 (Fig. S9†). We have performed control photochemical water oxidation experiments under nearly identical conditions, but using copper(II) chloride instead of POMs as a WOC. When 8 μM CuCl2·2H2O is used, water oxidation is observed with TON < 3. Albeit there is a limited number of reported studies devoted to investigation of photochemical water oxidation using homogeneous Cu complexes,19–21 to the best of our knowledge, the compounds 2 and 3 exhibit relatively high catalytic efficiency (Table 2). Specifically, their TOF values are three to five times higher compared to the pentacopper tungstosilicate analogue.21
As copper-based oxide materials are known for their catalytic activity in water oxidation,42 the oxides 2-ox and 3-ox obtained by thermal decomposition of 2 and 3, respectively, were also studied as heterogeneous catalysts in the similar ruthenium dye photo-oxidant system. When 0.1 mg of 2-ox or 3-ox were used, evolution of 65 nmol and 105 nmol of oxygen was observed, respectively (Fig. 8). As expected, 3-ox features significantly higher surface area of 13.5 m2 g−1 comparing to 5.5 m2 g−1 for 2-ox as was found from absorption studies (Fig. S10†).
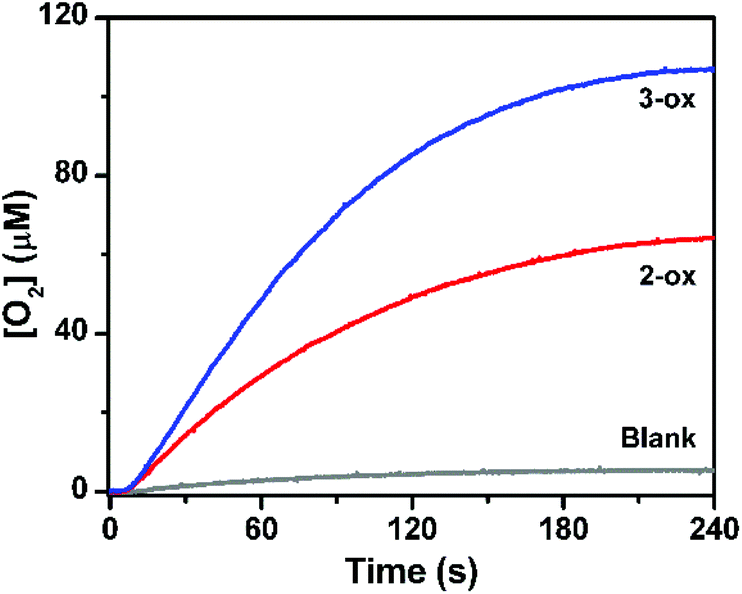 | ||
| Fig. 8 Oxygen evolution traces obtained in the heterogeneous water oxidation assay using 2-ox and 3-ox (0.1 mg each) as catalysts, [Ru(bpy)3](ClO4)2 (0.1 mM) as sensitizer, and Na2S2O8 (2 mM) as electron acceptor at pH 8.0. The photocatalysis was driven by blue light (3 W LEDs, λ = 450 nm). The blank trace is shown in grey. | ||
Electrochemical properties of 2 and 3
High catalytic efficiency of 2 and 3 in photochemical oxygen evolution inspired us to investigate their activity in electrochemical oxidation of water. The cyclic voltammograms (CV) of 2 and 3 were carried out in 0.04 M borate buffer at pH 8.0, thus, under similar conditions used in the photocatalytic assay. For both compounds, two anodic peaks were observed (Fig. 9). The first one-electron oxidative process appeared at +1.2 V versus NHE (normal hydrogen electrode) and could be assigned to the CuIII/CuII oxidation.16c,43 This was followed by a multi-electron oxidative process starting at +1.32 V versus NHE corresponding to water oxidation. The onset of the catalytic wave emerged at an overpotential of 0.56 V, which is comparable to the related copper-based water oxidation catalysts.16d,43,44 For this catalytic process, the peak current Ip is linearly dependent on the square root of the scan rate (Fig. 9, insets) that indicates a homogeneous process in solution, but not on the electrode surface. The difference in the catalytic performance of 2 and 3 may be also explained by the earlier onset of the pre-catalytic CuIII/CuII process for 3, so that the high-valent copper intermediate is thermodynamically more accessible by [Ru(bpy)3]3+ (E1/2 = +1.26 V versus NHE) that is generated from the light-excited triplet state of [Ru(bpy)3]2+ by persulfate. Since formation of the CuIII species is accompanied by the deprotonation of the coordinated water molecule, for both 2 and 3, the CuIII/CuII wave is non-reversible similarly to other water oxidation catalysts.16c,43,44b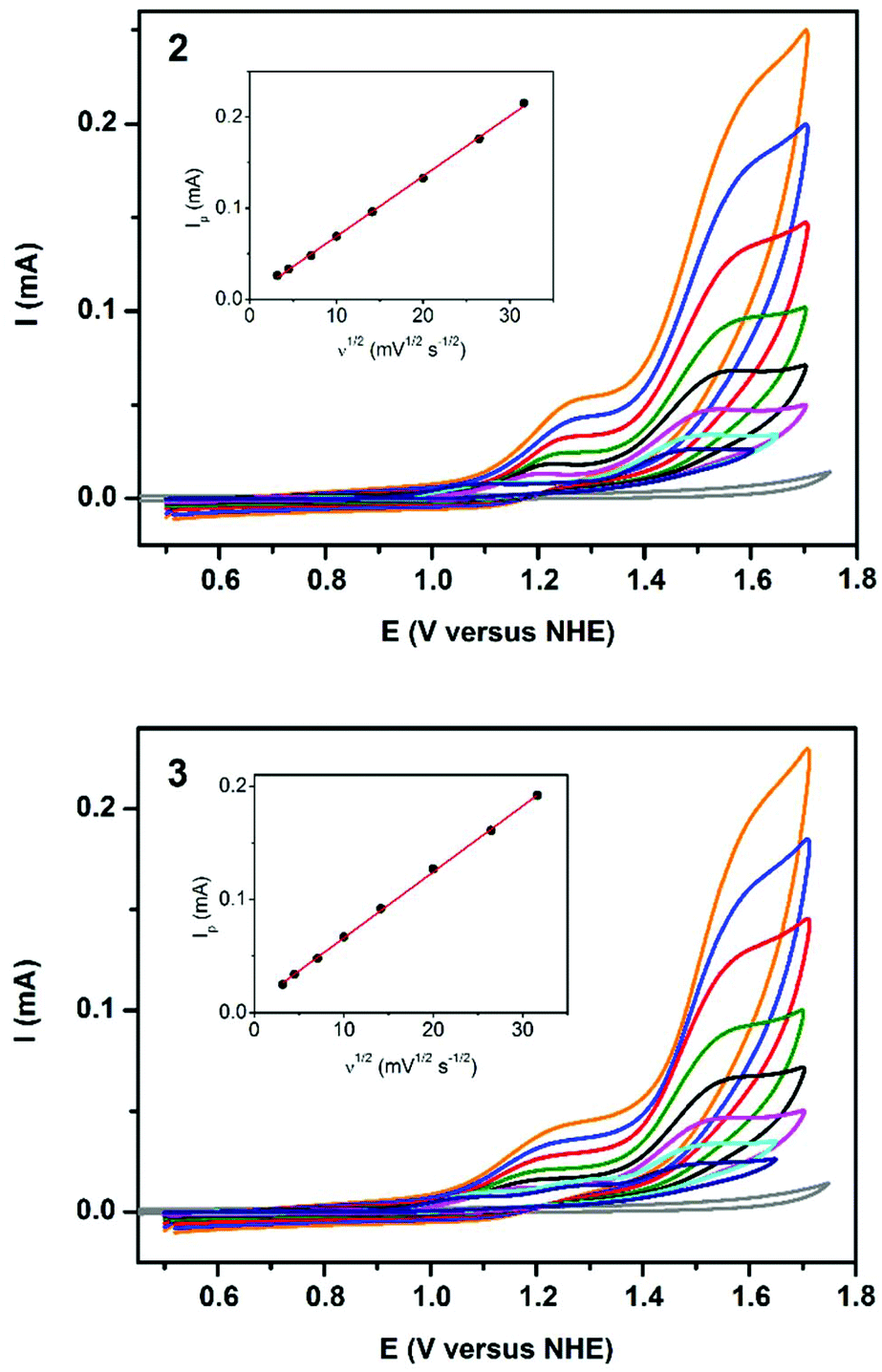 | ||
| Fig. 9 Cyclic voltammograms of 2 (top) and 3 (bottom) recorded in buffered (pH = 8.0) aqueous solutions at scan rates ν from 10 to 1000 mV s−1, and background recorded at ν = 100 mV s−1 (grey line). Insets: anodic current at E = 1.58 V versus NHE as function of ν1/2. | ||
Conclusions
In this study, three novel hybrid compounds constructed from the rare vanadomolybdate POMs and copper complex fragments with polyamine ligands have been described. The main difference in the complexes is various environment of copper leading to substantial change in catalytic properties. Compound 1 bearing two en molecules coordinated to Cu centre has been found to be inactive as the metal's coordination surrounding is fully saturated. At the same time, compounds 2 and 3 with one dien ligand per copper centre have proven to efficiently oxidize water to dioxygen in the photo-oxidant system driven by visible light at pH 8.0. Their catalytic performance relies on the relatively mild water oxidation overpotential in combination with their high stability under catalytic conditions. DLS experiments indicate that the catalytically active species 2 and 3 are molecular in nature, and presumptive in situ formation of copper hydroxide nanoparticles does not contribute to the oxygen evolution. The remarkable difference in the catalytic activity of isomers 2 and 3 has been attributed to the different position of the coordinated water molecule in respect to the vacant coordination site available for the binding of the second molecule of the substrate. Moreover, these structural peculiarities have further influence on formation of oxide materials by thermal decomposition of the complexes, especially, on the content of copper-based bronze. These findings together with synthetic availability of hybrid compounds based on POMs provide a promising guideline to design efficient copper-based WOCs. The relevant studies are currently ongoing.Experimental
Materials and general methods
All reagents were of analytical reagent grade and were used without further purification unless otherwise stated. CHN-analyses were performed using a Vario El Cube elemental analyzer. Infrared (IR) spectra were recorded in the range of 400–4000 cm−1 in KBr pellets using a PerkinElmer Spectrum BX spectrometer. Thermogravimetric analysis (TG/DTA) was performed in air using a TG-DTA system Setaram SETSYS 16/18. The TG/DTA curves were registered at a heating rate of 5 K min−1 in the temperature range 20–700 °C. Powder X-ray diffraction (XRD) patterns were collected in θ–2θ mode using a Shimadzu XRD-6000 diffractometer with Cu Kα radiation (λ = 1.5418 Å, graphite monochromator, 35 kV, 30 mÅ). The experiment was carried out in reflectance mode for the well-powdered sample in the flat glass container. Data collection has been performed in step scan mode (2θ step 0.02θ) at a scanning rate 2 s per point. The EPR spectra were recorded on Bruker ELEXSYS E500 CW-EPR spectrometer operating at X-band equipped with frequency counter and NMR Teslameter.Single-crystal X-ray crystallography
Details of the data collection and processing, structure solving and refinement are summarized in Table 3.| # | 1 | 2 | 3 |
|---|---|---|---|
| Empirical formula | C6H42CuMo6N8O30V2 (1) | C8H48Cu2Mo6N8O33V2 (2) | C8H54Cu2Mo6N8O36V2 (3) |
| Formula weight | 1447.53 | 1589.14 | 1643.19 |
| Crystal system | Triclinic | Orthorhombic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pccn | P121/n1 |
| a (Å) | 8.5468(5) | 11.8618(8) | 11.2569(3) |
| b (Å) | 10.9507(5) | 23.2484(12) | 9.1947(2) |
| c (Å) | 10.9914(4) | 15.7977(8) | 21.9132(5) |
| α (°) | 102.267(4) | 90 | 90 |
| β (°) | 112.333(5) | 90 | 99.328(2) |
| γ (°) | 99.415(5) | 90 | 90 |
| V (Å3) | 895.21(8) | 4356.5(4) | 2238.11(9) |
| Z | 1 | 4 | 2 |
| D calc (g cm−3) | 2.685 | 2.423 | 2.438 |
| μ(MoKα), mm−1 | 3.218 | 3.132 | 3.057 |
| F(000) | 701 | 3088 | 1604 |
| T/K | 298 | 298 | 298 |
| θ range (°) | 3.7–30.0 | 3.1–27.5 | 3.6–27.5 |
| Reflections collected/unique | 9455/5609 | 30081/5929 |
19449/7174 |
| R int | 0.0448 | 0.1797 | 0.0397 |
| Reflections with F > 4s(F) | 3921 | 2707 | 5746 |
| Parameters | 262 | 295 | 286 |
| R 1[F2 > 2σ(F2)] | 0.0445 | 0.0626 | 0.0419 |
| wR2 (all data) | 0.0711 | 0.1247 | 0.1050 |
| GOF on F2 | 0.934 | 0.954 | 1.046 |
| CCDC number | 1882180 | 1882181 | 1882182 |
Diffraction data for crystals of 1–3 were collected on an Xcalibur-3 diffractometer (MoKα radiation, λ = 0.71073 Å,CCD-detector, graphite monochromator, ω-scanning). Empirical absorption correction was provided with a multi-scan method using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm of the CrysAlisPro program package.45 Structures were solved by direct methods and refined against F2 within anisotropic approximation for all non-hydrogen atoms using the OLEX246 program package with SHELXT and SHELXL modules.47
In 1, oxygen atom of disordered water molecule (O15A, O15B) was restrained to be approximately isotropic to within 0.01 Å2. In 2, the same restrain was applied to all crystallization water molecules, to within 0.02–0.03 Å2.
Syntheses of the complexes
Method 2. Initially two different aqueous solutions were used for synthesis from copper salt. Solution A (10 ml) was prepared from V2O5 (0.109 g, 0.599 mmol) and (NH4)6Mo7O24·4H2O (0.847 g, 0.685 mmol) while solution B (5 mL) was obtained by mixing CuSO4·5H2O (0.100 g, 0.40 mmol) and dien (0.043 mL, 0.40 mmol). The solution A was heated up to 60–65 °C and kept stirring for 30 min with following addition of solution B. The reaction mixture was kept stirring for the next 30 minutes. The resulting solution (pH = 5.76) was filtered hot. The small quantity of green powder formed after cooling down to the room temperature was filtered in 2–3 hours. Green crystals of 2 were collected and air-dried in a day. Yield 0.09 g (28% by copper). After removal of 2, the supernatant was left for further evaporation yielding violet crystals of 3 in about two weeks. Yield 0.05 g (15% by copper).
Anal. calc. for C8H48Cu2Mo6N8O33V2 (2): C, 6.05; H, 3.04; N, 7.05. Found: C, 6.01; H, 3.06; N, 6.95%. IR of 2 (KBr, cm−1): 3440 (br), 3308 (m), 3260 (m), 3220 (br), 3153 (br), 3035 (br), 2960 (w), 2886 (w), 1685 (sh), 1650 (sh), 1605 (m), 1575 (m), 1466 (sh), 1425 (m), 1395 (sh), 1365 (sh), 1261 (w), 1145 (w), 1135 (w), 1150 (sh), 1090 (m), 1052 (sh), 1030 (m), 996 (sh), 942 (s), 912 (sh), 893 (vs), 871 (sh), 770 (vs), 700 (sh), 635 (m), 580 (m), 526 (m).
Anal. calc. for C8H54Cu2Mo6N8O36V2 (3): C, 5.85; H, 3.31; N, 6.82. Found: C, 5.93; H, 3.16; N, 7.02%. IR of 3 (KBr, cm−1): 3410 (br), 3332 (w), 3305 (w), 3214 (w), 3157 (br), 3084 (sh), 2955 (w), 2888 (w), 1618 (w), 1575 (w), 1465 (sh), 1426 (br), 1324 (w), 1300 (w), 1260 (w), 1236 (w), 1140 (w), 1095 (w), 1130 (w), 986 (w), 931 (m), 863 (vs), 773 (sh), 755 (br, s), 714 (sh), 621 (m), 579 (m), 529 (m).
Synthesis of oxide phases 2-ox and 3-ox
Thermal degradation of complexes 2, 3 was performed under air atmosphere, the prepared samples being denoted as 2-ox and 3-ox. The samples were heated to 550 °C at a heating rate of 5 °C min−1, held at this temperature for one hour and allowed to cool in air.Oxygen evolution experiments
Oxygen evolution was detected polarographically by using a standard Clark-type oxygraph electrode. The electrode was separated from the sample solution by a Teflon membrane, and the signal was recorded every 0.1 s by using the Oxygraph software package. Air-saturated aqueous solution ([O2]20°C = 276μm) was used for calibration of the electrode. The cell was purged with argon gas before each experiment, and the solution in the cell (1 ml) was continuously stirred. Visible light LEDs (λ = 450nm, 3 W) were used as illumination sources. Prior to water oxidation studies, the concentrations of photosensitizer [Ru(bpy)3]2+ and sacrificial electron acceptor S2O82– were varied to optimize the reaction conditions. Under optimized conditions, the concentrations of the components in the catalytic systems in the Clark cell were as follows: 4 μm of WOC, 0.1 mM of {[Ru(bpy)3](ClO4)2}·6H2O, 2 mM of Na2S2O8 in borate buffer (0.04 M, pH = 8.0). For studies of heterogeneous water oxidation with mixed oxides as catalysts, 0.1 mg of 2-ox or 3-ox, 0.1 mM of {[Ru(bpy)3](ClO4)2}·6H2O, and 2 mM of Na2S2O8 in borate buffer (0.04 mM, pH = 8.0) were used. All oxygen evolution experiments were repeated three times each to afford reproducible turnover frequency (TOF) and turnover number (TON) values. The TOF and TON were calculated based on the total number of copper centres in compounds. TOF = number of O2 molecules evolved per second per one Cu ion at the maximum rate of oxygen evolution (mol(O2) mol(Cu)−1 s−1); TON = total number of O2 molecules per one Cu atom evolved by the moment of time, when reaction rate decreased to zero (mol(O2) mol(Co)−1).
Electrochemistry
Cyclic voltammetry was carried out by using an Autolab potentiostat with a GPES electrochemistry interface (Eco Chemie). A three-electrode system with a 3 mm glass carbon working electrode, a glassy carbon rod counter electrode, and an Ag/AgCl in saturated KCl aqueous solution reference electrode (E = +0.195 versus normal hydrogen electrode) was used. The counter and reference electrodes were in compartments separated from the bulk solution by fritted disks. The working electrode was polished with 0.05 μm alumina paste and sonicated before use. The solution under study (5 ml) contained 2 or 3 (0.25 mM), borate buffer (0.04 M, pH 8.0) and KClO4 (0.1 M) as a supporting electrolyte. Before all measurements, oxygen was removed from the cell by bubbling argon through the stirred solutions. Samples were kept under argon during the measurements.Adsorption studies
N2 adsorption isotherms were measured using an Accelerated Surface Area and Porosimetry System ASAP 2060 at 77 K (liquid nitrogen bath). Samples (35 mg each) were activated under dynamic vacuum (10−4 Pa) using a Micromeritic SmartVacPrep sample preparation unit. Helium gas was used for the determination of the cold and warm free space of the sample tubes. The surface area was calculated from the adsorption isotherms using Micrometrics ASAP 2060 Software (v. 1.00).Dynamic light scattering studies
DLS experiments were performed by using a Zetasizer Nano S scattering system (Malvern Instruments Ltd) that used a uni-phase He–Ne laser (633 nm; 4 mW) working in cross auto-correlation mode. The scattering angle was set to 90° in respect the incident laser. The intensity correlation curves were analyzed with the Zetasizer family software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SIS gratefully thanks Dr Gustav Berggren for kind hosting. The work was partially supported by the Swedish Institute (Grant No. 23913/2017) and the Ministry of Education and Science of Ukraine. The authors thank Dr O. Bieda for assistance in powder XRD data analysis.References
- (a) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed; (b) L. Hammarström, Acc. Chem. Res., 2015, 48, 840 CrossRef PubMed.
- (a) M. Yamamoto, J. Föhlinger, J. Petersson, L. Hammarström and H. Imahori, Angew. Chem., Int. Ed., 2017, 56, 3329 CrossRef CAS PubMed; (b) F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86 RSC; (c) D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006 CrossRef CAS PubMed.
- For selected reviews on artificial photosynthesis, see: (a) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed; (b) H. Zhou, R. Yan, D. Zhang and T. Fan, Chem. – Eur. J., 2016, 22, 9870 CrossRef CAS; (c) D. R. Ort, S. S. Merchant, J. Alric, A. Barkan, R. E. Blankenship, R. Bock, R. Croce, M. R. Hanson, J. M. Hibberd, S. P. Long, T. A. Moore, J. Moroney, K. K. Niyogi, M. A. J. Parry, P. P. Peralta-Yahya, R. C. Prince, K. E. Redding, M. H. Spalding, K. J. van Wijk, W. F. J. Vermaas, S. von Caemmerer, A. P. M. Weber, T. O. Yeates, J. S. Yuan and X. G. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8529 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26 CrossRef CAS.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef CAS.
- (a) C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS; (b) F. A. Frame, T. K. Townsend, R. L. Chamousis, E. M. Sabio, T. Dittrich, N. D. Browning and F. E. Osterloh, J. Am. Chem. Soc., 2011, 133, 7264 CrossRef CAS; (c) T. Audichon, T. W. Napporn, C. Canaff, C. Morais, C. Comminges and K. B. Kokoh, J. Phys. Chem. C, 2016, 120, 2562 CrossRef CAS.
- W. Smith, J. Power Sources, 2000, 86, 74 CrossRef CAS.
- (a) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS PubMed; (b) L. Duan, C. M. Araujo, M. S. G. Ahlquist and L. Sun, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15584 CrossRef CAS PubMed; (c) L. Wang, L. Duan, Y. Wang, M. S. G. Ahlquist and L. Sun, Chem. Commun., 2014, 50, 12947 RSC; (d) J. M. Thomsen, D. L. Huang, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2015, 44, 12452 RSC.
- (a) A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607 CrossRef CAS; (b) M. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421 RSC.
- (a) M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886 CrossRef CAS PubMed; (b) J. Ma, Q. Wang, W.-L. Man, H.-K. Kwong, C.-C. Ko and T.-C. Lau, Angew. Chem., Int. Ed., 2015, 54, 5246 ( Angew. Chem. , 2015 , 127 , 5335 ) CrossRef PubMed; (c) B. Schwarz, J. Forster, M. K. Goetz, D. Yücel, C. Berger, T. Jacob and C. Streb, Angew. Chem., Int. Ed., 2016, 55, 6329 ( Angew. Chem. , 2016 , 128 , 6437 ) CrossRef CAS PubMed.
- (a) W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS PubMed; (b) Z. Codolà, I. Garcia-Bosch, F. Acuña-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 8042 CrossRef PubMed; (c) J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807 CrossRef CAS PubMed; (d) C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273 CrossRef CAS PubMed; (e) S. I. Shylin, M. V. Pavliuk, L. D'Amario, F. Mamedov, J. Sá, G. Berggren and I. O. Fritsky, Chem. Commun., 2019, 55, 3335 RSC.
- (a) D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579 CrossRef CAS PubMed; (b) D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178 CrossRef CAS PubMed; (c) J.-W. Wang, P. Sahoo and T.-B. Lu, ACS Catal., 2016, 6, 5062 CrossRef CAS.
- (a) M. Zhang, M. Zhang, C. Hou, Z. Ke and T. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042 ( Angew. Chem. , 2014 , 126 , 13258 ) CrossRef CAS PubMed; (b) J.-W. Wang, W.-L. Liu, D.-C. Zhong and T.-B. Lu, Coord. Chem. Rev., 2019, 378, 237 CrossRef CAS.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed.
- (a) M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048 CrossRef CAS PubMed; (b) D. M. Ryan, M. K. Coggins, J. J. Concepcion, D. L. Ashford, Z. Fang, L. Alibabaei, D. Ma, T. J. Meyer and M. L. Waters, Inorg. Chem., 2014, 53, 8120 CrossRef CAS PubMed.
- (a) T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273 CrossRef CAS PubMed; (b) D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689 CrossRef CAS PubMed; (c) M. K. Coggins, M.-T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226 ( Angew. Chem. , 2014 , 126 , 12422 ) CrossRef CAS PubMed; (d) S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586 CrossRef CAS PubMed.
- (a) K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384 CrossRef CAS; (b) B. Rudshteyn, K. J. Fisher, H. M. C. Lant, K. R. Yang, B. Q. Mercado, G. W. Brudvig, R. H. Crabtree and V. S. Batista, ACS Catal., 2018, 8, 7952 CrossRef CAS.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Bernet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758 CrossRef CAS PubMed.
- R.-J. Xiang, H.-Y. Wang, Z.-J. Xin, C.-B. Li, Y.-X. Lu, X.-W. Gao, H.-M. Sun and R. Cao, Chem. – Eur. J., 2016, 22, 1602 CrossRef CAS PubMed.
- R. Terao, T. Nakazono, A. R. Parent and K. Sakai, ChemPlusChem, 2016, 81, 1064 CrossRef CAS.
- L. Yu, X. Du, Y. Ding, H. Chen and P. Zhou, Chem. Commun., 2015, 51, 17443 RSC.
- (a) L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef; (b) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 ( Angew. Chem. , 2010 , 122 , 1780–1803 ) CrossRef CAS PubMed; (c) N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed.
- (a) D. E. Katsoulis, Chem. Rev., 1998, 98, 359 CrossRef CAS PubMed; (b) E. Coronado and C. J. Gomez-Garcia, Chem. Rev., 1998, 98, 273 CrossRef CAS PubMed.
- (a) Q.-S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS PubMed; (b) J. Wei, Y.-Y. Feng, P.-P. Zhou, Y. Liu, J.-Y. Xu, R. Xiang, Y. Ding and C.-C. Zhao, ChemSusChem, 2015, 8, 2630 CrossRef CAS PubMed; (c) H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268 CrossRef CAS PubMed; (d) X.-B. Han, Y.-G. Li, Z.-M. Zhang, H.-Q. Tan, Y. Lu and E.-B. Wang, J. Am. Chem. Soc., 2015, 137, 5486 CrossRef CAS PubMed; (e) Q. Han and Y. Ding, Dalton Trans., 2018, 47, 8180 RSC.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed.
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217 CrossRef CAS.
- (a) M. V. Pavliuk, V. G. Makhankova, O. V. Khavryuchenko, V. N. Kokozay, I. V. Omelchenko, O. V. Shishkin and J. Jezierska, Polyhedron, 2014, 81, 597 CrossRef CAS; (b) H. I. Buvailo, V. G. Makhankova, V. N. Kokozay, I. V. Zatovsky, I. V. Omelchenko, S. V. Shishkina, P. Zabierowski, D. Matoga and J. Jezierska, Eur. J. Inorg. Chem., 2016, 5456 CrossRef CAS; (c) H. I. Buvailo, V. G. Makhankova, V. N. Kokozay, I. V. Omelchenko, S. V. Shishkina, P. Zabierowski, D. Matoga and J. Jezierska, Eur. J. Inorg. Chem., 2017, 3525 CrossRef CAS.
- (a) M. V. Pavliuk, E. Mijangos, V. G. Makhankova, V. N. Kokozay, S. Pullen, J. Liu, J. Zhu, S. Styring and A. Thapper, ChemSusChem, 2016, 9, 2957 CrossRef CAS PubMed; (b) M. V. Pavliuk, V. G. Makhankova, V. N. Kokozay, I. V. Omelchenko, J. Jezierska, A. Thapper and S. Styring, Polyhedron, 2015, 88, 81 CrossRef CAS.
- D. S. Nesterov, O. V. Nesterova, V. N. Kokozay and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2014, 4496 CrossRef CAS.
- (a) J. Fuchs and H. Hartl, Angew. Chem., Int. Ed. Engl., 1976, 15, 375 CrossRef; (b) A. J. Bridgeman and G. Cavigliasso, Inorg. Chem., 2002, 41, 3500 CrossRef CAS PubMed.
- (a) Z. L. Li, Y. Wang, L. C. Zhang, J. P. Wang, W. S. You and Z. M. Zhu, Dalton Trans., 2014, 43, 5840 RSC; (b) J. P. Wang, H. X. Ma, L. C. Zhang, W. S. You and Z. M. Zhu, Dalton Trans., 2014, 43, 17172 RSC; (c) X. M. Li, Y. G. Chen, C. Su, S. Zhou, Q. Tang and T. Shi, Inorg. Chem., 2013, 52, 11422 CrossRef CAS PubMed.
- (a) E. Burkholder, S. Wright, V. Golub, C. J. O'Connor and J. Zubieta, Inorg. Chem., 2003, 42, 7460 CrossRef CAS PubMed; (b) C. Y. Sun, Y. G. Li, E. B. Wang, D. R. Xiao, H. Y. An and L. Xu, Inorg. Chem., 2007, 46, 1563 CrossRef CAS PubMed.
- A. Björnberg, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1995 CrossRef.
- (a) J. Wang, J. Wang, P. Ma and J. Niu, J. Coord. Chem., 2009, 62, 2641 CrossRef CAS; (b) J. Wang, Y. Feng, J. Zhao, P. Ma, X. Zhang and J. Niu, J. Coord. Chem., 2009, 62, 3754 CrossRef CAS; (c) J. Li, L. Xu, N. Jiang, L. Zhao and F. Li, Struct. Chem., 2011, 22, 1339 CrossRef CAS.
- (a) T. Lu, X. Li, Z. Mao, W. Qiu, L. Ji and K. Yu, Polyhedron, 1998, 17, 75 CrossRef CAS; (b) E. E. Martsinko, L. Kh. Minacheva, V. S. Sergienko, E. A. Chebanenko and I. I. Seifullina, Russ. J. Coord. Chem., 2010, 55, 1874 CAS.
- Angular structural index parameter τ can be calculated as τ = (β − α)/60°, where α and β are the two largest valence angles of the coordination polyhedron, α < β. A. W. Addison and T. N. Rao, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- A. B. P. Lever, Inorganic electronic spectroscopy, 2nd edn, Elsevier, New York, 1984 Search PubMed.
- (a) S. Kumara, R. P. Sharma, P. Venugopalan, M. Witwicki and V. Ferrettic, J. Mol. Struct., 2016, 1123, 124–132 CrossRef; (b) B. Kurzak, D. Kroczewska and J. Jezierska, Polyhedron, 1998, 17, 1831 CrossRef CAS.
- (a) V. K. Kato, E. Takayama-Muromachi and Y. Kanke, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 1841 CrossRef; (b) C. J. Patridge, C. Jaye, H. Zhang, A. C. Marschilok, D. A. Fischer, E. S. Takeuchi and S. Banerjee, Inorg. Chem., 2009, 48, 3145 CrossRef CAS PubMed.
- (a) Z. Chen and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 700 ( Angew. Chem. , 2013 , 125 , 728 ) CrossRef CAS PubMed; (b) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013 CrossRef CAS PubMed; (c) M. C. Kafentzi, R. Papadakis, F. Gennarini, A. Kochem, O. Iranzo, Y. Le Mest, N. Le Poul, T. Tron, B. Faure, A. J. Simaan and M. Reglier, Chem. – Eur. J., 2018, 24, 5213 CrossRef CAS PubMed.
- (a) F. Yu, F. Li, B. Zhang, H. Li and L. Sun, ACS Catal., 2015, 5, 627 CrossRef CAS; (b) T. N. Huan, G. Rousse, S. Zanna, I. T. Lucas, X. Xu, N. Menguy, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2017, 56, 4792–4796 ( Angew. Chem. , 2017 , 129 , 4870 ) CrossRef CAS PubMed.
- J. Shen, M. Wang, J. Gao, H. Han, H. Liu and L. Sun, ChemSusChem, 2017, 10, 4581 CrossRef CAS PubMed.
- (a) M. M. Najafpour, S. Mehrabani, Y. Mousazade and M. Hołyńska, Dalton Trans., 2018, 47, 9021–9029 RSC; (b) S. Nestke, E. Ronge and I. Siewert, Dalton Trans., 2018, 47, 10737 RSC; (c) J. Shen, M. Wang, P. Zhang, J. Jiang and L. Sun, Chem. Commun., 2017, 53, 4374 RSC.
- CrysAlis PRO, Agilent Technologies, 2014 Search PubMed.
- O. Dolomanov, L. Bourhis, R. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1882180–1882182. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi00040b |
| This journal is © the Partner Organisations 2019 |