
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting photochemical activity by Ni doping of mesoporous CoO nanoparticle assemblies†
Georgia
Velegraki
a,
Ioannis
Vamvasakis
a,
Ioannis T.
Papadas
b,
Sotiris
Tsatsos
c,
Anastasia
Pournara
d,
Manolis J.
Manos
 d,
Stelios A.
Choulis
c,
Stella
Kennou
c,
Georgios
Kopidakis
a and
Gerasimos S.
Armatas
*a
d,
Stelios A.
Choulis
c,
Stella
Kennou
c,
Georgios
Kopidakis
a and
Gerasimos S.
Armatas
*a
aUniversity of Crete, Department of Materials Science and Technology, Heraklion 71003, Greece. E-mail: garmatas@materials.uoc.gr
bCyprus University of Technology, Department of Mechanical Engineering and Materials Science and Engineering, Limassol 3041, Cyprus
cUniversity of Patras, Department of Chemical Engineering, Patra 26504, Greece
dUniversity of Ioannina, Department of Chemistry, Ioannina 45110, Greece
First published on 4th February 2019
Abstract
The rational design of semiconductor nanostructures is of utmost importance for efficient solar energy conversion and environmental remediation. In this article, we report high-surface-area mesoporous networks consisting of Ni-implanted cubic CoO (Co1−xNixO) nanoparticles as promising catalysts for the detoxification of aqueous Cr(VI) solutions. Mechanistic studies with X-ray photoelectron, UV–vis optical absorption, fluorescence and electrochemical impedance spectroscopy and theoretical (DFT) calculations indicate that the performance enhancement of these catalysts arises from the high charge transfer kinetics and oxidation efficiency of surface-reaching holes. By tuning the chemical composition, the Co1−xNixO mesoporous catalyst at 2 atomic% Ni content imparts outstanding photocatalytic Cr(VI) reduction and water oxidation activity, corresponding to an apparent quantum yield (QY) of 1.5% at λ = 375 nm irradiation light.
1. Introduction
Hexavalent chromium (Cr(VI)) constitutes a serious chemical residue in wastewater arising from various industrial activities, such as electroplating, leather tanning and paint manufacturing among others. Contamination by Cr(VI) species results in groundwater pollution with devastating effects on the environment.1 In particular, Cr(VI) is a non-biodegradable contaminant that is responsible for human carcinogens and mutagens according to the International Agency for Research on Cancer (IARC), classifying it as a Group 1 carcinogen with a lethal dose (LD50) of 50–150 mg kg−1 for an adult.2 Therefore, neutralization of Cr(VI)-bearing solutions is of utmost importance in terms of protecting the environment and human health.Recently, the research community has strived for the remediation of Cr(VI) ions through the photocatalytic reduction method, that is, reduction of Cr(VI) to less harmful Cr(III), which can be precipitated as Cr(OH)3 or Cr2O3 in alkaline solutions.3 To this end, various semiconductor materials, such as TiO2, ZnO, WO3, SnS2, Bi2S3 and CdS, have been investigated as catalysts for Cr(VI) photoreduction.4 Nevertheless, although promising, these materials suffer from low solar energy absorption, short lifetime of photogenerated electron–hole pairs and poor electrical conductivity, which adversely impact their viability for use in photocatalysis. Furthermore, most of these catalysts operate in the presence of a hole scavenger such as ascorbic acid, citric acid and ethylenediamine-tetraacetic acid (EDTA), which seriously impedes their utilization in Cr(VI)-rich wastewaters. Therefore, devising cost-effective catalysts for detoxification of Cr(VI)-contaminated aquatic systems is an ongoing research challenge.
Cobalt monoxide (CoO) has recently emerged as an attractive material for redox catalysis, gas sensor devices and lithium-ion batteries due to its high redox activity, visible light response (band gap (Eg) ∼2.2–2.8 eV) and chemical stability.5 CoO crystallizes in a cubic rock-salt and hexagonal wurtzite structure. Generally, the cubic close-packed structure of CoO is thermodynamically more stable than the hexagonal ones but suffers from poor catalytic activity.6 Catalytic systems of cubic CoO have only recently demonstrated promising activity for CO oxidation and photocatalytic water-splitting reactions.7
Herein, we report for the first time chemically stable and robust mesoporous networks of Ni-implanted cubic CoO nanoparticles (NPs) that present high photocatalytic activity for Cr(VI) reduction and water oxidation under UV and visible light irradiation. We utilize a polymer-assisted self-assembly of colloidal NPs to assemble mesoporous Co1−xNixO (x = 0–0.05) NP-linked frameworks with high interstitial porosity. This method utilizes the NPs as building block units and is based on the coassembly of these nanoscopic particles with organic structure-directing agents to construct three-dimensional structures with open-pore morphology.8 The resulting materials, unlike individual NPs and bulk microstructures, feature a nanometer-sized framework perforated by uniform pores, which allows rapid mass transport and provides plenty of exposed surface-active sites available for reaction. Moreover, we highlight the effect of the Ni dopant on the electronic structure and photochemical performance of CoO by using a combination of experiment and theoretical methods. The results of this study can offer new insights into the design and development of efficient photocatalysts for environmental remediation and energy conversion applications.
2. Results and discussion
2.1 Synthesis and structural analysis of mesoporous Co1−xNixO assemblies
The synthesis of mesoporous assemblies from colloidal Co1−xNixO NPs (denoted as Co1−xNixO MNAs, x = 0, 0.01, 0.02 and 0.05) was accomplished by a polymer-templated self-assembly method. In short, a poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) block copolymer was utilized to template the aggregating self-assembly of BF4−-stabilized Co1−xNixO NPs in a mixed N,N-dimethyloformamide (DMF)/tetrahydrofuran (THF) solution. The reaction mixture was kept under stirring for 2 h approximately, followed by slow evaporation of the solvent (within 5–6 days) at 40 °C in order for mesostructured NP/polymer composites to be formed. Finally, the polymer template was removed by decomposition at 350 °C to give a continuous network of connected NPs with a large internal surface area (details are given in the Experimental section). The elimination of the organic phase upon heating was confirmed by thermogravimetric analysis (TGA), see Fig. S1 and S2.† The Ni composition in implanted samples was determined by energy dispersive X-ray spectroscopy (EDS) and anodic stripping voltammetry (ASV). Results showed that the molar ratio Ni/Co in the samples was very close to the expected composition from the stoichiometry of reactions (Table 1).| Sample | Ni contenta (atomic%) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | |
|---|---|---|---|---|---|
| EDS | ASV | ||||
| a Ni/(Co + Ni) atomic ratio based on EDS and anodic stripping voltammetry (ASV) analysis. | |||||
| CoO MNAs | 137 | 0.15 | 4.4 | ||
| Co0.99Ni0.01O | 0.99 | 1.09 ± 0.14 | 108 | 0.16 | 4.3 |
| Co0.98Ni0.02O | 1.89 | 2.06 ± 0.07 | 112 | 0.16 | 4.5 |
| Co0.95Ni0.05O | 4.81 | 4.97 ± 0.44 | 115 | 0.15 | 4.4 |
X-ray diffraction (XRD) measurements were performed to investigate the crystal structure of the as-prepared materials. In Fig. 1a, all samples exhibit broad but distinct XRD peaks that correspond to the cubic rock-salt structure of CoO with Fm3m symmetry (JCPDS no 48-1719). No impurity peaks corresponding to other chemical phases of cobalt like Co3O4 were detected, indicating the phase purity of the materials. However, due to the low concentration of Ni in Co1−xNixO MNAs, no corresponding XRD peaks can be observed. Using the peak width of the (200) diffraction and Scherrer's equation, the crystallite size of the mesoporous materials was estimated to be ∼6.5–7 nm, which is very close to the grain size of the precursor NPs (data not shown).
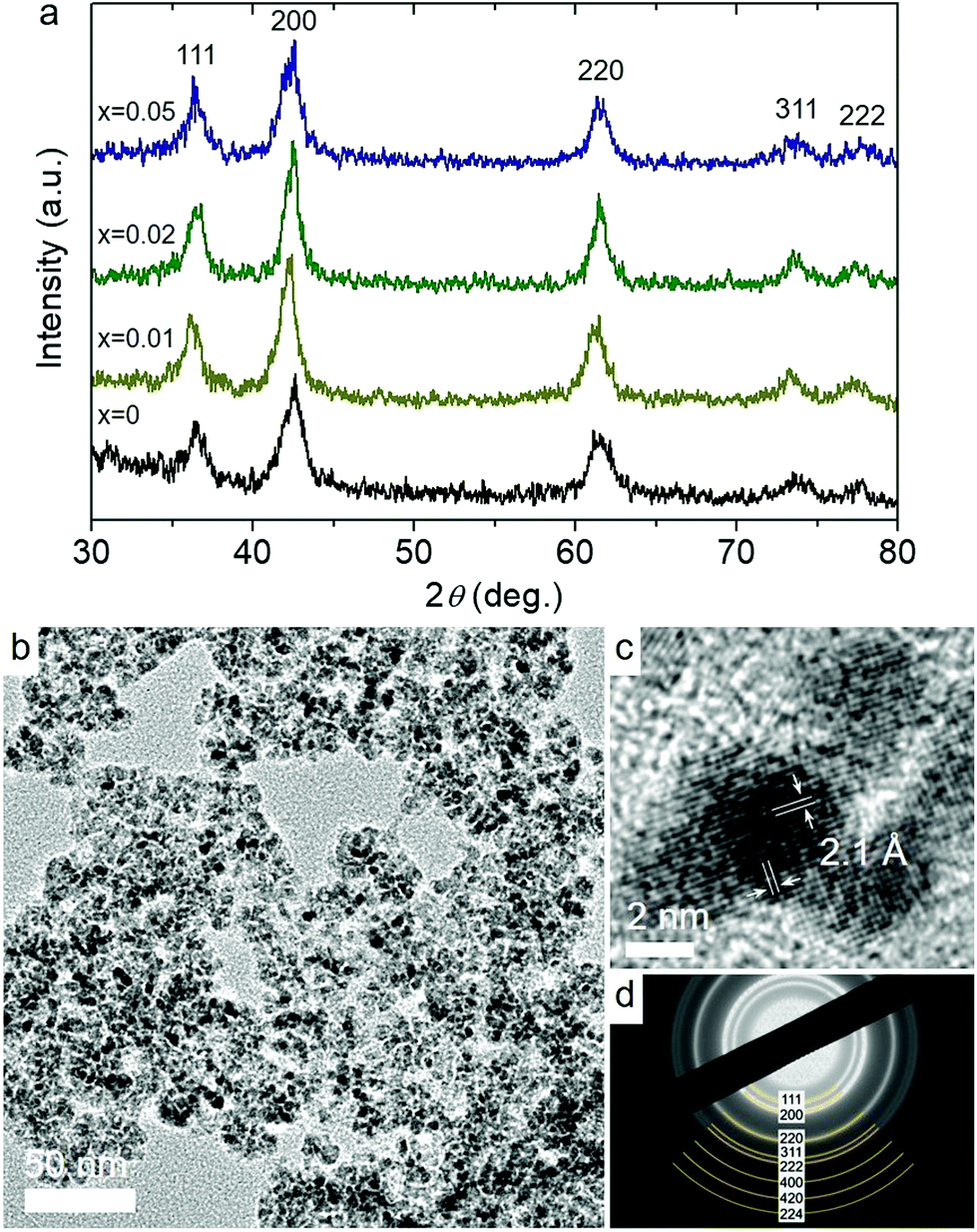 | ||
| Fig. 1 (a) XRD patterns of Co1−xNixO MNAs. (b) Typical TEM image, (c) high-resolution TEM, showing the lattice fringes of CoO along the [001] direction, and (d) SAED pattern of Co0.98Ni0.02O MNAs. | ||
Fig. 1b and Fig. S3a† display typical transmission electron microscopy (TEM) images obtained from mesoporous assemblies of CoO and Co0.98Ni0.02O NPs; Co0.98Ni0.02O MNAs are the most active catalyst among the studied materials. The images show a porous network of connected NPs with an average size of ca. 5–6 nm, which is very close to the crystallite sizes obtained from XRD, implying minimal grain coarsening of NPs during synthesis. Crystal structure features of CoO and Co0.98Ni0.02O MNAs were also characterized by high-resolution TEM (HRTEM) and selected-area electron diffraction (SAED). The HRTEM images shown in Fig. 1c and the inset of Fig. S3a† suggest single-crystalline NPs, showing distinct lattice fringes throughout the entire particles with ∼2.1 Å interlayer distances that correspond to the (200) crystallographic planes of cubic CoO. Besides, the SAED patterns in Fig. 1d and Fig. S3b† signify the polycrystalline nature of the porous frameworks, displaying a series of Debye-Scherrer rings. In agreement with XRD results, all the diffraction rings can be assigned to the cubic phase of CoO.
Nitrogen physisorption experiments corroborate that Co1−xNixO MNAs contain mesopores within the assembled structure. All samples showed type-IV adsorption–desorption isotherms with a H3 type hysteresis loop at high relative pressures (Fig. S4†), which are characteristic of mesoporous solids with slit-like pores.9 The specific surface areas, which were derived using the Brunauer–Emmett–Teller (BET) method on the absorption data, and the total pore volumes of the Ni-implanted samples were found to be 108–115 m2 g−1 and 0.15–0.16 cm3 g−1, respectively, which are comparable to those of pure CoO MNAs (137 m2 g−1 and 0.15 cm3 g−1, respectively). The slightly lower surface area of the Ni-doped samples relative to the unmodified CoO MNAs could be attributed to the close packing of the Co1−xNixO NPs which form a more dense (crack-free) pore wall, although the slightly higher mass density of the NiOx phase (6.67 g cm−3) compared to that of CoO (6.44 g cm−3) is a possible explanation. Analysis of the adsorption data using the non-local density functional theory (NLDFT) method revealed a narrow size distribution of pores with the pore diameter being in the range of 4.3–4.5 nm. All the analytical and textural properties of Co1−xNixO MNAs are listed in Table 1.
The surface chemical state of pristine CoO and Co0.98Ni0.02O MNA materials was examined by X-ray photoelectron spectroscopy (XPS). For the CoO MNA catalyst, the Co 2p3/2 XPS core-level peak is detected at a binding energy of 781.1 eV with a shake-up satellite feature at about 786.4 eV, providing convincing evidence of the existence of Co(II) ions (Fig. 2a).10 The Co 2p3/2 signal is clearly observed at slightly lower binding energies (780.8 eV) for the Co0.98Ni0.02O MNAs, possibly due to a partial electron transfer from Ni to Co through the bridging oxygen atoms.11 Specifically, the single occupied 3d t2g orbital of high-spin Co(II) (t2g5eg2 electron configuration) can interact with the oxygen lone pairs, accepting electrons via π-donation. After Ni doping, the fully occupied 3d t2g states of Ni(II) (t2g6eg2 electron configuration) can strengthen the partial electron transfer from oxygen to Co(II) due to the repulsive interactions with the occupied 2p orbitals of oxygen.12 In agreement with this, the O 1s XPS spectrum of Co0.98Ni0.02O MNAs, which is deconvoluted into two peaks assigned to the lattice oxygen (530.0 eV) and the hydroxyl groups (532.0 eV), is slightly red-shifted compared to that of CoO (530.3 eV and 532.2 eV), Fig. 2b. This downshift in binding energies (by ∼0.2–0.3 eV) for oxygen atoms is thought to be related to the incorporation of Ni. Comparatively, the O 1s spectrum of the polycrystalline NiO sample shows two distinct peaks at 529.4 eV and 531.2 eV (Fig. S5†). Since the Ni content in Co0.98Ni0.02O MNAs is as low as 1.56 wt%, it is difficult to directly detect by XPS (the XPS Ni 2p3/2 peak is typically observed in the range of 854.2–854.5 eV). In line with XPS results, the valence band (VB) spectra of CoO and Co0.98Ni0.02O MNAs provide an additional hint for the incorporation of Ni into the CoO lattice (Fig. 2c). The CoO shows a photoemission onset, which is defined as the energetic difference between the Fermi energy and the VB edge, at ca. 0.31 eV, indicating a p-type character. For the Co0.98Ni0.02O MNAs, the photoemission onset is shifted to ca. 0.26 eV, reflecting a downshift of the Fermi level due to the Ni doping. Consistent with this interpretation, the photoelectron emission onset of polycrystalline NiO was found at 0.22 eV.
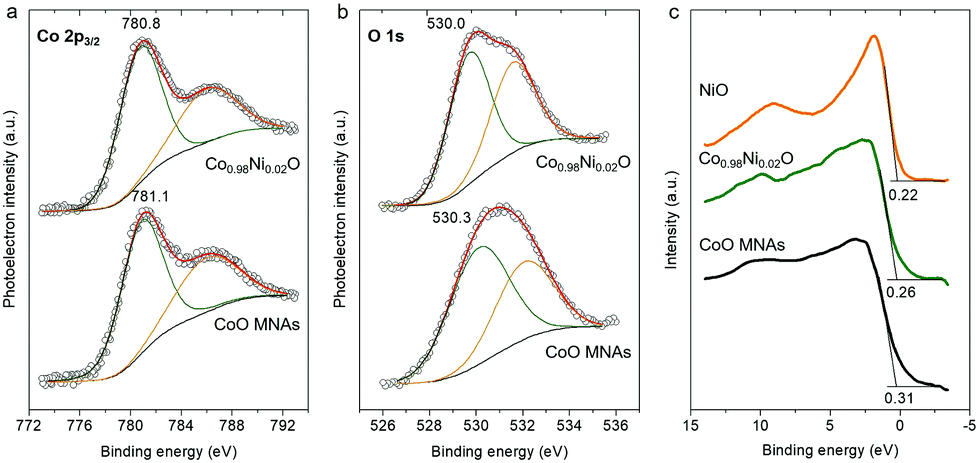 | ||
| Fig. 2 XPS spectra of the (a) Co 2p3/2, (b) O 1s and (c) valence band region of CoO and Co0.98Ni0.02O MNAs. In panel (c), the XPS valence band spectrum of polycrystalline NiO is also given. | ||
2.2 Photocatalytic Cr(VI) reduction activity
The catalytic activity of the title materials was investigated in photoreduction of aqueous Cr(VI) solutions under λ >360 nm light irradiation. Fig. 3a depicts the evolution of Cr(VI) reduction over Co1−xNixO MNA catalysts with different Ni loadings (from 0 to 5 atomic%). The comparison shows that Co0.98Ni0.02O MNAs exhibit better performance for Cr(VI) photoreduction, achieving ∼90% Cr(VI) conversion in 4 h, which is markedly higher than that of pure CoO MNAs (∼46% Cr(VI) conversion level, 4 h). The outstanding photocatalytic performance of Co0.98Ni0.02O MNAs is also evident from their high apparent quantum yield (QY ∼1.5% at 375 nm and ∼0.8% at 410 nm), which is comparable or even higher to that of other highly active catalysts, such as hexagonal CoO NP assemblies (∼1.61% at 375 nm),13 Au/TiO2-Pt (∼1.0% at 550 nm),14 CuFe2O4/CdS (∼1.1% at 430 nm)15 and CuAl2O4/TiO2 (∼0.11% under white light)16 composites. Of note, the optimum catalyst dosage was found to be 0.3 g L−1 by performing a series of photocatalytic Cr(VI) reduction tests with different catalyst concentrations (from 0.2 to 0.4 g L−1, see Fig. S6†). Moreover, because of the visible light absorption of Co1−xNixO NPs (Eg ∼2.5–2.7 eV, as estimated by optical absorption spectra, see the inset of Fig. 5), Co1−xNixO MNAs can also carry out photocatalytic Cr(VI) reduction with reasonable activity under λ >420 nm irradiation. As shown in Fig. 3a, ∼27% and ∼50% Cr(VI) conversion yield was obtained with visible-light irradiated CoO and Co0.98Ni0.02O MNA dispersions in 4 h, respectively.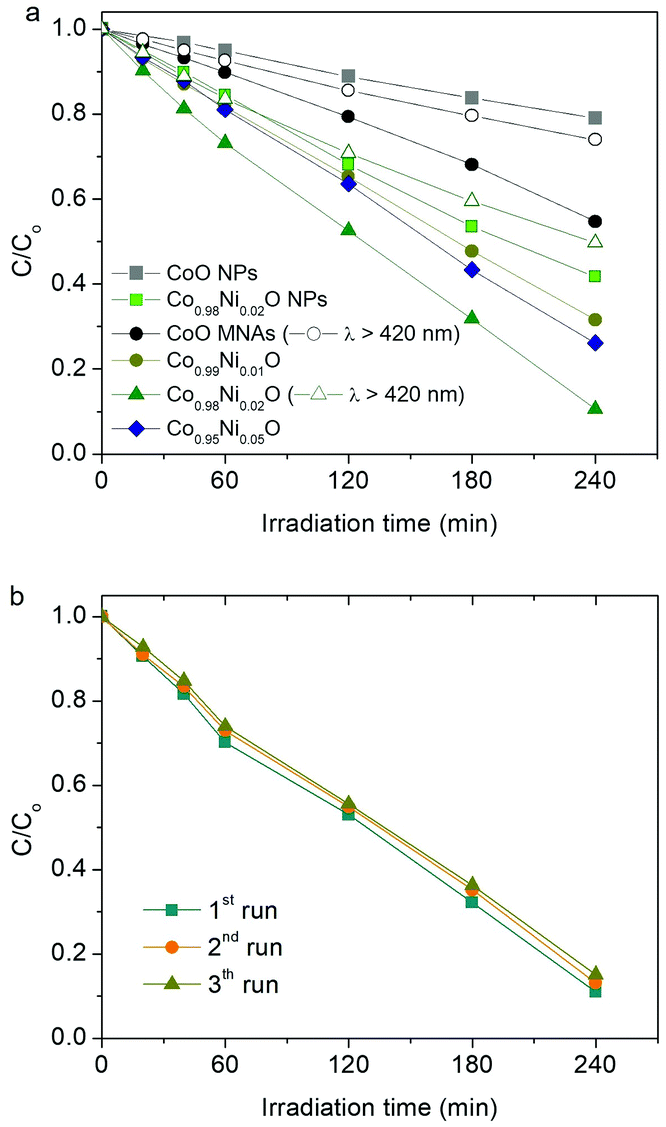 | ||
| Fig. 3 (a) Photocatalytic reduction of aqueous Cr(VI) with Co1−xNixO MNAs catalysts. The photocatalytic activity of isolated CoO and Co0.98Ni0.02O NPs under λ >360 nm light and CoO and Co0.98Ni0.02O MNA catalysts under λ >420 nm light irradiation is also given. (b) Recycling study of Co0.98Ni0.02O MNAs. Reaction conditions: 0.3 g L−1 catalyst, 50 mg L−1 Cr(VI) solution, pH = 2, λ >360 nm light, 20 °C. | ||
In addition to the chemical composition, morphological effects may also contribute to the high photocatalytic activity of Co1−xNixO MNAs. As a proof of concept, we investigated the photocatalytic Cr(VI) reduction activity of isolated (BF4−-capped) CoO and Co0.98Ni0.02O NPs under identical conditions and the results are depicted in Fig. 3a. It can be seen that individual CoO and Co0.98Ni0.02O NPs afforded a considerably lower catalytic activity (ca. 21% and 58% Cr(VI) conversion level in 4 h, respectively) than that obtained for Cr(VI) photoreduction over the corresponding mesoporous NP assemblies. These results clearly indicate that the material's open-pore structure and accessible surface area play an important role in the reduction rate of Cr(VI).
The Co0.98Ni0.02O MNA catalyst also demonstrates very good stability over a period of 12 h. The reusability of the catalyst was assessed within three recycling experiments, in which the catalyst after each run was collected by centrifugation and re-dispersed in a fresh Cr(VI) aqueous solution. As shown in Fig. 3b, the Co0.98Ni0.02O MNAs retain more than 94% of their initial activity after recycling. Moreover, XRD, N2 porosimetry and XPS characterization of the reused sample indicated that its crystal structure, porous morphology and surface chemical state are well maintained after catalysis, substantiating high durability (see Fig. S7–S9†).
2.3 Effect of the Ni dopant on photochemical activity
To understand the oxidation mechanism over Co1−xNixO assemblies, we carried out photoreduction of Cr(VI) (50 mg L−1) in an air-tight quartz cell and the evolved oxygen gas was analyzed by gas chromatography. The results showed that Co0.98Ni0.02O MNAs are effective at oxidizing water to dioxygen (Fig. 4), giving an average O2 evolution rate of ∼3.5 μmol h−1 after 4 h of irradiation. Moreover, to prove that the generation of oxygen is indeed a photoinduced effect, the evolved gas was monitored with an on-line gas analyzer. When light on and off conditions were applied to a Co0.98Ni0.02O MNA dispersion (0.3 g L−1) for photocatalytic Cr(VI) reduction, a transient O2 evolution response was detected (Fig. S10†), suggesting that the present Cr(VI) photoreduction proceeds simultaneously with the water oxidation reaction. Thus, the overall reaction can be described by the following equation in which the hydrogen chromate (HCrO4−) ions are the predominating species in conditions with pH <6 and Cr(VI) concentrations <100 mg L−1.17 | (1) |
 | ||
| Fig. 4 Comparison of photocatalytic Cr(VI) reduction (black) and oxygen evolution (experimental: blue line, calculated from the HCrO4− concentration-time data and stoichiometry of reaction (1): red line) as a function of irradiation time for the Co0.98Ni0.02O MNA catalyst under λ >360 nm light irradiation (Reaction conditions: 0.3 g L−1 catalyst, 50 mg L−1 Cr(VI) solution, λ >360 nm light, pH = 2, 20 °C). | ||
It is noteworthy that the calculated oxygen evolution derived from eqn (1) matches well with the experimental results (Fig. 4). In particular, we obtained a HCrO4− reduction rate of about 5.2 μmol h−1 which, based on eqn (1), consists of a ∼3.9 μmol h−1 O2 evolution rate. These results prove that, under UV–visible irradiation, a prominent fraction of surface-reaching holes is associated with the water oxidation reaction to produce oxygen.
Apart from the oxygen evolution, photooxidation of surface hydroxyl (–OH) groups to hydroxyl radicals (˙OH) is a possible option. Recently, we have demonstrated that this is a viable process for hexagonal CoO catalysts in the photocatalytic reduction of Cr(VI).13 To elucidate this possibility, we resort to the fluorescence (FL) study of the Cr(VI) photoreduction over pure and Ni-doped CoO (2% Ni loading) MNAs in the presence of coumarin as a fluorophore; coumarin reacts with hydroxyl radicals to produce fluorescence umbelliferone. The time evolution of the FL emission of umbelliferone reveals that the production rate of ˙OH radicals, defined as the ratio of the intensity of umbelliferone to the intensity of coumarin, is comparable between these two materials (see Fig. S11†). Also, the FL signal of umbelliferone does not show an obvious intensity increase after 1 h of illumination. Therefore, it is reasonable to conclude that the observed OHaq− oxidation process for both pure and Ni-doped CoO MNA catalysts is minimal and, instead, the oxidation product is almost exclusively O2. This is consistent with the O2 evolution data shown in Fig. 4.
The above catalytic results clearly suggest that the four-electron water oxidation process (2H2O → O2 + 4H+ + 4e−) at the Co1−xNixO surface dictates the overall reaction rate. As a proof of concept, the Cr(VI) reduction activity of Co0.98Ni0.02O MNAs is also investigated in the presence of phenol, citric acid and EDTA as sacrificial electron donors. Since oxidation of these organic compounds is thermodynamically and kinetically more favorable than water oxidation (Eox for phenol, citric acid, EDTA and water is 0.97 V, 1.2 V, 1.17 V and 0.82 V vs. NHE (pH 7), respectively),18 it is anticipated that this process will enhance the kinetics of the Cr(VI) reduction. As expected, the Cr(VI) degradation is significantly accelerated with the addition of 1 equiv. of the above sacrificial reagents, resulting in complete conversion of Cr(VI) in 3–3.5 h (Fig. S12a†). Assuming that the reaction rate is proportional to the Cr(VI) concentration, the photocatalytic reaction can be described by the first-order kinetics of the Langmuir–Hinshelwood model: ln(C/C0) = −kt, where C0 and C are the concentration of Cr(VI) at initial time and time t, respectively, and k is the apparent reaction rate constant. Thus, kinetic analysis of the temporal evolution of the Cr(VI) concentration reveals that photoreduction proceeds at a rate (k) of 4.4 × 10−3 min−1, 7.0 × 10−3 min−1 and 9.7 × 10−3 min−1 with phenol, citric acid and EDTA, respectively (Fig. S12b†). In comparison, an about two to six times lower reaction rate (1.7 × 10−3 min−1) was observed without a sacrificial reagent, signifying that the presence of organic contaminants in the Cr(VI)-containing wastewater has an additive effect on improving the photocatalytic activity of Co1−xNixO MNAs.
To elucidate the effect of the Ni dopant on the photocatalytic activity of CoO NPs, the electrochemical behavior of the as-prepared materials was delineated by electrochemical impedance spectroscopy (EIS). The Mott–Schottky plots (1/Csc2vs. E) of the Co1−xNixO MNA depositions onto a fluorine-doped tin oxide (FTO) substrate recorded in a 0.5 M Na2SO4 electrolyte (pH 7) are shown in Fig. 5. Using extrapolation to 1/Csc2 = 0, the flat-band potentials (EFBs) calculated for Co1−xNixO MNAs were 1.15 (x = 0), 1.17 (x = 0.01), 1.20 (x = 0.02) and 1.22 (x = 0.05) V versus the normal hydrogen electrode (NHE). Consistent with VB XPS results, the negative slope of the 1/Csc2–E curves confirms the p-type behavior of the catalysts. The anodic shift in EFB for CoO upon Ni doping is due to the formation of Co–O–Ni linkages in implanted Co1−xNixO samples. In fact, substitution of Co(II) with Ni(II) impurities can create localized Ni 3d states near the VB maximum, increasing the hole density in Co1−xNixO. The presence of these acceptor states will shift the Fermi level downward, improving the p-type conductivity of CoO, as suggested by VB XPS measurements and theoretical density functional theory (DFT+U) calculations. Electronic structure calculations show hybridization of O 2p and Co/Ni 3d states and direct transitions. The projected electronic density of states (DOS) for pure CoO in the rocksalt structure near the Fermi level (Fig. 6a) gives an electronic band gap of 2.0 eV (the quasiparticle bandgap value is higher and can be found with GW calculations that are beyond the scope of this work). In order to examine the effects of doping on the electronic structure, Co atoms were replaced by Ni atoms in the CoO simulation cells. DFT+U calculations for Co0.5Ni0.5O show that the separation between valence and conduction band edges increases by 0.1 eV and that there is strong mixing of the O 2p and Co/Ni 3d orbitals (Fig. 6b). These trends are also confirmed by calculations for smaller Ni concentrations with larger simulation cells (Co0.75Ni0.25O and Co0.9375Ni0.0625O). Consistent with theoretical calculations, the carrier (acceptor) density (NA) of Co1−xNixO MNAs, as calculated by the slope of the 1/Csc2–E plots, increases from 3.31 × 1016 to 4.57 × 1016 cm−3 with a higher level of nickel doping (Table S1†).
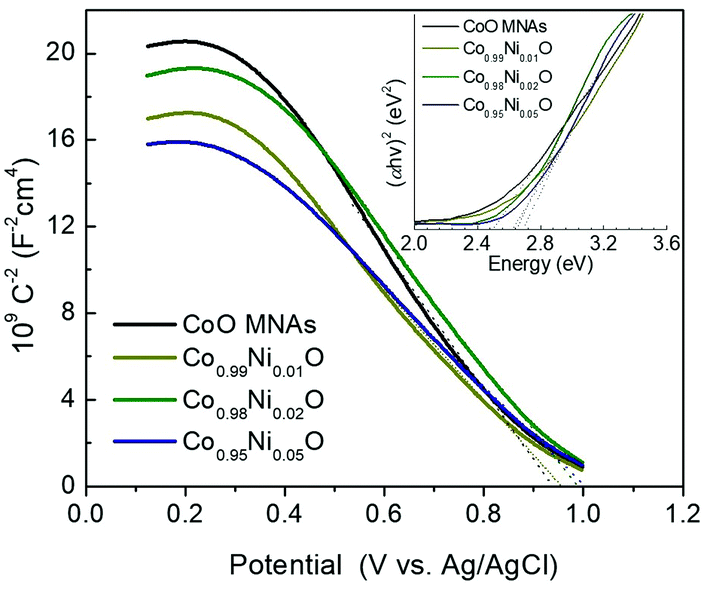 | ||
| Fig. 5 Mott–Schottky plots (Inset: Tauc plots derived from UV–vis diffuse reflectance spectra) of the Co1−xNixO MNA catalysts. | ||
 | ||
| Fig. 6 Projected DOS for (a) CoO and (b) Co0.5Ni0.5O rocksalt crystal structures. The DOS profiles near the Fermi level give an energy band gap of 2.0 and 2.1 eV for CoO and the Co0.5Ni0.5O lattice, respectively. | ||
In Fig. 7a, the energy band diagram for each catalyst is illustrated. For heavily p-doped semiconductors such as CoO, it is quite reasonable to assume that the EFB level lies very close to the VB edge. Thus, the position of the conduction band (CB) edge can be obtained by the equation ECB = EFB + Eg. UV–vis diffuse reflectance spectroscopy was used to obtain optical absorption spectra of Co1−xNixO MNAs, from which the Eg values were estimated to be 2.50, 2.63, 2.66 and 2.69 eV for CoO and Ni-implanted samples with 1%, 2% and 5% Ni content, respectively (Table S1†), using Tauc plot analysis [(Fhv)2versus photon energy, where F, h, and v are the Kubelka–Munk function of the reflectance, Planck constant, and light frequency, respectively] (inset of Fig. 5). As shown in Fig. 7a, while a small perturbation of the CB edge is observed, the VB edge position of Co1−xNixO MNAs shifts gradually to the anodic direction with increasing Ni doping, making them better electron acceptors for water oxidation, thus signifying superior Cr(VI) photoreduction kinetics.
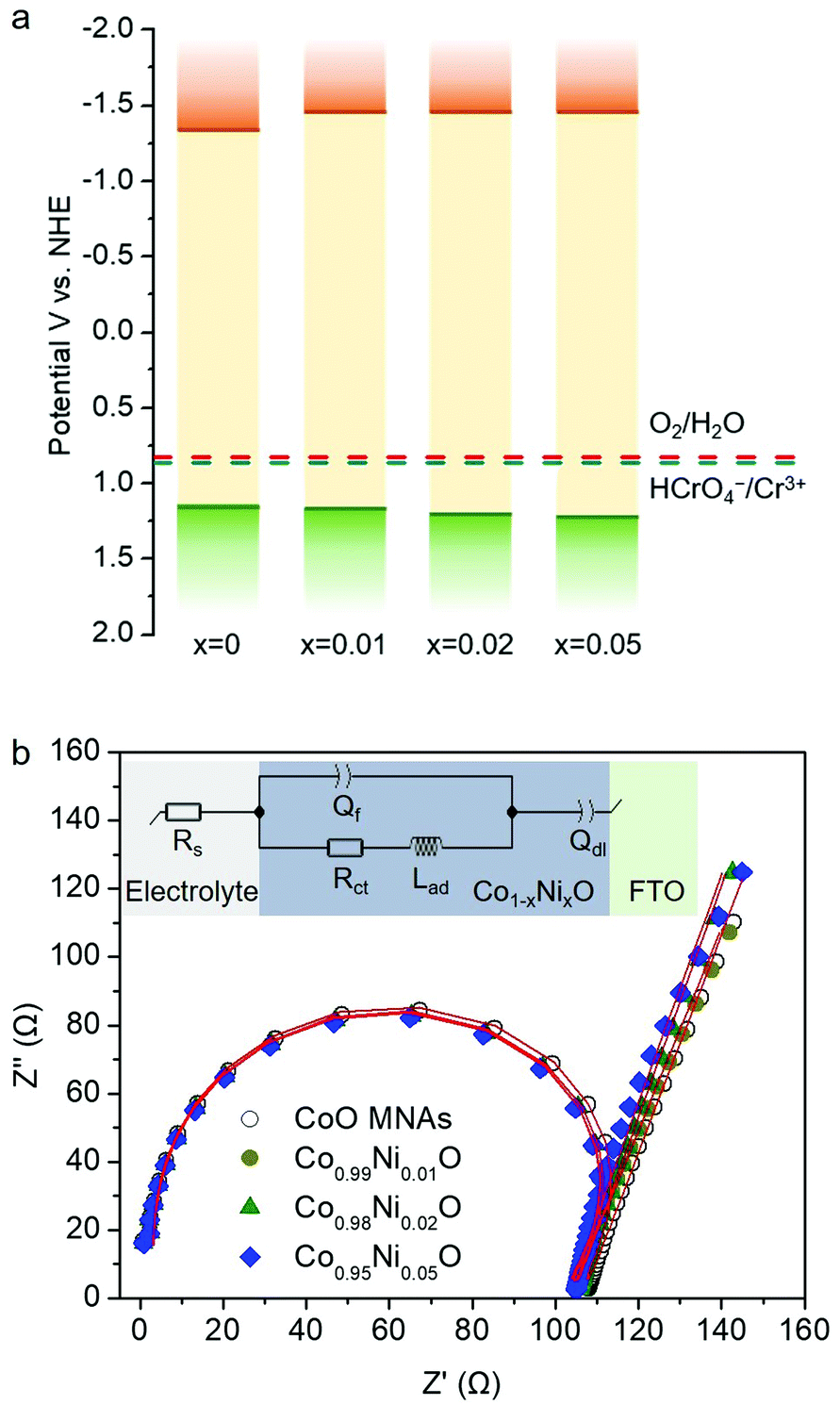 | ||
| Fig. 7 (a) Energy band diagrams and (b) Nyquist plots of the Co1−xNixO MNAs. | ||
In addition, from EIS measurements and Nyquist Plots, we received information about the charge-transfer resistance (Rct) for each catalyst (Fig. 7b). The EIS measurements were carried out in a conventional three-electrode cell containing 0.5 M KOH solution in the frequency range from 1 Hz to 1 MHz. An equivalent circuit model Rs(Qf/(RctLad)Qdl) (see the inset of Fig. 7b) was used to simulate the EIS curve of the fabricated Co1−xNixO MNA electrodes. In this circuit model, Rs represents the electrolyte resistance, Rct is the charge-transfer resistance, and Qf and Qdl elements account for the defect resistance (pores, cracks and grain boundaries) of the solid film and the double layer capacitance, respectively. In addition, an inductor (Lad), regarding the pseudo-inductive behaviour observed in the high frequency domain (often caused by disordered charge-carrier relaxation and disordered movement of redox species at the surface of electrode),19 was also necessary for fitting the experimental results. Apparently, results of this study revealed that Ni-doped samples possess a lower Rct than pure CoO MNAs does, affirming better charge carrier conductivity, which correlates well with their high Cr(VI) photoreduction activity. In particular, a Rct value of 103.8, 100.7, 100.5 and 99.8 Ω is obtained for pure and 1%, 2% and 5% Ni implanted CoO MNAs (Table S2†). In fact, a lower Rct value is anticipated to enhance the photoactivity of the catalyst by facilitating faster transport of charge carriers within the framework and prolonging the lifetime of photogenerated electron–hole pairs. Therefore, compared with CoO MNAs, the higher performance of Ni-doped samples can be attributed to the enhanced charge transfer and oxidative ability of surface-reaching holes. In line with this, the Co0.98Ni0.02O MNA catalyst exhibits higher mobility of the photogenerated carriers according to the photocurrent density (Jph) measurements; the Jph for Co1−xNixO MNAs was found to be 9.52 (x = 0), 11.01 (x = 0.01), 19.89 (x = 0.02) and 14.36 (x = 0.05) mA cm−2 under 420–760 nm irradiation at 1 V applied bias (see details in the Experimental section). Thus, the improved photochemical properties of Co0.98Ni0.02O MNAs can be interpreted as a result of the high p-type conductivity and efficient dissociation of excitons into spatially separated electron–hole pairs.
3. Conclusions
In summary, high-surface-area Ni-doped CoO NP assemblies have been successfully prepared by a polymer-assisted self-assembly method, and then used as photocatalysts for the detoxification of Cr(VI) aqueous solutions. Significantly, the Co1−xNixO MNA catalyst at 2 atomic% Ni content achieves higher Cr(VI) photoreduction and water oxidation activity (>90% Cr(VI) conversion and ∼3.5 μmol h−1 O2 evolution rate after 4 h of irradiation) with 1.5% apparent QY at 375 nm, while demonstrating very good stability, outperforming previous results. Mechanistic studies with X-ray photoelectron, UV–vis optical absorption, fluorescence and electrochemical impedance spectroscopy experiments and theoretical (DFT) calculations reveal that Ni doping of CoO causes a gradual shift of the VB edge position to the anodic direction and enhancement of the p-type conductivity of CoO. These electronic characteristics effectively improve the charge-transfer process and oxidation efficiency of surface-reaching holes for water oxidation. Such Ni-implanted CoO NP assemblies manifest improved photochemical properties and demonstrate good possibilities for widespread uses in photocatalysis and environmental remediation.4. Experimental
4.1 Synthesis of Co1−xNixO NPs
Co1−xNixO (x = 0, 0.01, 0.02 and 0.05) NPs were prepared according to a modified literature procedure.20 For a typical synthesis of Co0.98Ni0.02O NPs, 1.47 mmol of Co(acac)3 (98%, Sigma-Aldrich), 0.03 mmol of Ni(NO3)2·6H2O (98%, Alfa Aesar) and 300 mmol of oleylamine (70%, Sigma-Aldrich) were heated at 135 °C under an inert atmosphere for 5 h. The resulting brown solution was then slowly heated to 200 °C with a heating rate of ∼2 °C min−1 and maintained at this temperature for 3 h, giving a brown suspension. Next, the supernatant was removed by centrifugation, retrieving the Co0.98Ni0.02O NPs in a powdery form. The Co0.98Ni0.02O NPs were then dispersed in hexane (95%, Sigma-Aldrich) to form a stable colloidal solution (10 mg mL−1). A similar procedure was followed for the synthesis of 1% and 5% Ni-doped CoO NPs, utilizing 1.485 mmol and 0.015 mmol and 1.425 mmol and 0.075 mmol of Co(acac)3 and Ni(NO3)2·6H2O, respectively.4.2 Ligand exchange of Co1−xNixO NPs
A dispersion of Co1−xNixO NPs in hexane (6 mL) was added to a solution of NOBF4 (60 mg, 97%, Acros Organics) in DMF (6 mL, 99.9%, Sigma-Aldrich) and the mixture was stirred at room temperature until the NPs were transferred to the DMF phase. The BF4−-capped Co1−xNixO NPs were then collected by precipitation with toluene (99.7%, Sigma-Aldrich), followed by centrifugation. The ligand-stripped Co1−xNixO NPs were dispersed in DMF to form a stable colloidal solution of 120 mg mL−1.4.3 Synthesis of Co1−xNixO NP assemblies
Mesoporous Co1−xNixO nanoparticle assemblies (MNAs) were prepared following a previously reported procedure with some modifications.6 For a typical synthesis of mesoporous Co1−xNixO nanoparticle assemblies (MNAs), 0.2 g of block copolymer Pluronic F127 (OH(CH2CH2O)100(CH(CH3)CH2O)64(CH2CH2O)100H, Mn ∼12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich) were dissolved in 1.5 mL of tetrahydrofuran (THF, >99%, Sigma-Aldrich) with continuous stirring. To this solution, 0.5 mL of colloidal Co1−xNixO NP solution in DMF was added dropwise and the mixture was stirred at room temperature for 2 h. Then, the resulting mixture was placed in an oven at 40 °C and left for 5 days to give a mesostructured Co1−xNixO/polymer composite. The resulting dry powder was subsequently heated to 350 °C for 4 h under N2 flow (ca. 50 cm3 min−1), using a heating rate of 1 °C min−1, to yield the mesoporous product.
000, Sigma-Aldrich) were dissolved in 1.5 mL of tetrahydrofuran (THF, >99%, Sigma-Aldrich) with continuous stirring. To this solution, 0.5 mL of colloidal Co1−xNixO NP solution in DMF was added dropwise and the mixture was stirred at room temperature for 2 h. Then, the resulting mixture was placed in an oven at 40 °C and left for 5 days to give a mesostructured Co1−xNixO/polymer composite. The resulting dry powder was subsequently heated to 350 °C for 4 h under N2 flow (ca. 50 cm3 min−1), using a heating rate of 1 °C min−1, to yield the mesoporous product.
4.4 Photocatalytic reactions
In a typical procedure, a quantity of catalyst (10–20 mg) was dispersed in 50 ml of 50 mg L−1 Cr(VI) aqueous solution, which were prepared by dissolving K2Cr2O7 in deionized (DI) water. The solution pH was adjusted with dilute sulfuric acid. Before irradiation, the suspension was magnetically stirred in the dark for 30 min to ensure adsorption/desorption equilibrium. The photoreduction reaction was performed by irradiating the solution with UV–visible or visible light, using a 300 W Xe lamp (Variac Cermax) with cut-off filters allowing λ >360 nm or λ >420 nm, respectively. All the experiments were carried out at 20 ± 2 °C using a water bath cooling system. During reaction, the concentration of Cr(VI) in the solution was monitored via the 1,5-diphenylcarbazide (DPC) colorimetric method, using a PerkinElmer Lambda 25 UV–vis spectrometer. Control experiments were carried out with the addition of phenol, citric acid and EDTA (1 equiv.) into the reaction mixture.For the photooxidation of water to oxygen, 15 mg of the Co0.98Ni0.02O MNA catalyst and 50 mL of 50 mg L−1 Cr(VI) aqueous solution (pH = 2) were placed into a 100 mL airtight quartz tube. The temperature of the suspension was maintained at 20 ± 2 °C by using an external water-cooling system. The mixture was first purged with argon for 30 min under atmospheric pressure to remove any dissolved air and then irradiated with a 300 W Xe lamp (λ >360 nm). The O2 generated from the reaction was analyzed with a gas chromatograph (Shimadzu GC-2014) equipped with a thermal conductivity detector.
The O2 evolution transient experiments were performed under λ >360 nm light irradiation in an air-tight quartz cell containing 0.3 g L−1 Co0.98Ni0.02O MNA catalyst and 50 mg L−1 Cr(VI) aqueous solution (pH = 2) and the evolved gas was monitored with an on-line gas analyzer (Hiden HPR-20 QIC).
The apparent quantum yield (QY) of the catalyst was calculated by analyzing the amount of reduced Cr(VI) at given monochromatic irradiation, using LED light with wavelengths of 375 and 410 nm, respectively:
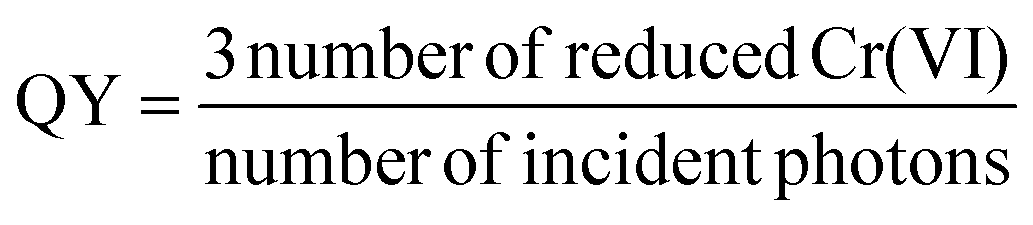 | (2) |
The flux of incident photons was measured using a StarLite energy meter equipped with a FL400A-BB-50 fan-cooled thermal sensor (Ophir Optronics Ltd, Jerusalem).
4.5 Physical characterization
Thermogravimetric analysis was performed on a PerkinElmer Diamond instrument under a N2 atmosphere (∼200 mL min−1 flow rate) with a heating rate of 5 °C min−1. Powder X-ray diffraction (XRD) patterns were collected on a PANalytical X'Pert Pro X-ray diffractometer (45 kV and 40 mA) using Cu Kα (λ = 1.5418 Å) radiation (45 kV, 40 mA). XRD data were recorded in the 2θ range of 30–80° with a 2θ step size of 0.01° and a scanning speed of 0.1° min−1 in Bragg–Brentano geometry. Elemental microprobe analyses were performed by using a JEOL JSM-6390LV scanning electron microscope (SEM) equipped with an Oxford INCA PentaFETx3 energy-dispersive X-ray spectroscopy (EDS) detector (Oxfordshire, UK). Data acquisition was performed at least five times for each sample using an accelerating voltage of 20 kV and a 100 s accumulation time. Anodic stripping voltammetry (ASV) was performed using a trace metal analyzer (797 VA Computrace, Metrohm AG Ltd, Switzerland). A three-electrode system was used comprising a hanging mercury drop electrode (HMDE) as the working electrode, platinum (Pt) as the auxiliary electrode and Ag/AgCl (3 M KCl) as the reference electrode. Before analysis, the Ni-doped CoO samples were dissolved in 5 mL aqua regia and the solutions were diluted appropriately, so that the measuring concentrations were less than 10 ppb. The analysis has been conducted following the AN-V-87 protocol developed by Metrohm.21 X-ray photoelectron spectroscopy (XPS) measurements were carried out using a mon-monochromatic Mg Kα line at 1253.6 eV (12 kV with 20 mA anode current) and a Leybold LH EA11 electron energy analyzer operated at a constant pass energy of 100 eV. The analyzed area was a 1.5 × 5 mm2 rectangle, placed near the center of the powder-covered area on each specimen surface. In all XPS spectra, the binding energy of the predominant aliphatic contribution to the C 1s peak at 284.8 eV was used as a reference. Transmission electron microscopy (TEM) images were taken with a JEOL JEM-2100 electron microscope equipped with a LaB6 filament and operated at 200 kV accelerating voltage. Samples were prepared by suspending fine powders in anhydrous ethanol and then drop casting on a copper grid covered with a carbon film. Nitrogen adsorption–desorption isotherms were measured at −196 °C on a Quantachrome NOVA 3200e sorption analyzer. Before measurement, samples were degassed at 100 °C under vacuum (<10−5 Torr) for at least 12 h. The specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) method on the adsorption data in the relative pressure range of 0.05–0.22. The total pore volumes were estimated from the adsorbed amount at the relative pressure (P/P0) of 0.98, and the pore size distributions were obtained from the adsorption branch of the isotherms, using the non-local density functional theory (NLDFT) method.22 UV–vis diffuse reflectance spectra were recorded with a PerkinElmer Lambda 950 optical spectrophotometer, using BaSO4 powder as a 100% reflectance standard. Diffuse reflectance data were converted to absorption using the Kubelka–Munk function F = (1 − R)2/(2R), where R is the measured reflectance.23 Fluorescence spectra were obtained at room temperature on a Lumina Fluorescence spectrometer (Thermo scientific) equipped with a 150 W Xe lamp.4.6 Electrochemical measurements
Mott–Schottky measurements were performed in a 0.5 M Na2SO4 aqueous electrolyte (pH = 7) using a Metrohm Autolab PGSTAT 302N potentiostat. A three-electrode set-up, with a platinum plate (1.0 × 2.0 cm2) and a silver–silver chloride (Ag/AgCl, 3 M KCl) as the counter and reference electrodes, respectively, was adopted to study the samples. The capacitance of the semiconductor/electrolyte interface was obtained at 1 kHz, with 10 mV AC voltage amplitude.For the semiconductor–electrolyte interface, the capacitance Csc of the space charge region can be described as follows:
 | (3) |
The carrier density (NA) of the samples can be calculated using the following equation:
 | (4) |
For Nyquist plots, a small AC perturbation of 20 mV was applied to the electrodes, and the different current output was measured throughout a frequency range of 1 to 106 Hz. The steady state DC bias was kept at 0 V (open-circuit potential) throughout the EIS experiments. All these experiments were conducted under dark conditions. The measured potential vs. the Ag/AgCl reference electrode was converted to the normal hydrogen electrode (NHE) scale using the formula: ENHE = EAg/AgCl + 0.210 V.
The working electrodes for impedance-potential measurements were fabricated as follows: ∼10 mg of each sample was dispersed in 1 mL DI water and the mixture was subjected to sonication in a water bath until a uniform suspension was formed. After that, 40 μL of the suspension was drop-cast onto the surface of the fluorine-doped tin oxide (FTO, 9 Ω sq−1) substrate, which was masked with an epoxy resin to expose an effective area of 1.0 × 1.0 cm2. The sample was dried in a 60 °C oven for 30 minutes.
4.7 Photoelectrochemical measurements
Photoelectrochemical measurements were carried out in an air-tight three-electrode cell with the samples as the working electrode (1 cm2 illumination area), Ag/AgCl (3 M KCl) as the reference electrode, and a platinum wire as the counter electrode. Photocurrent data were collected at a potential of 1 V (vs. Ag/AgCl) in 0.5 M KOH aqueous electrolyte. Before analysis, the electrolyte was purged with N2 for 30 min to remove the oxygen. The photocurrent densities of the samples were measured using a Metrohm Autolab PGSTAT 302N potentiostat coupled with a neutral white light-emitting diode (WLED, λ = 420–760 nm).4.8 Theoretical calculations
Density functional theory (DFT) calculations were performed using the Vienna ab initio Simulation Package (VASP).24 The projector augmented wave method (PAW) for the core electrons and the nuclei was used.25 For the exchange–correlation functional, we tested both the local density approximation (LDA)26 and the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE).27 We found that LDA describes the electronic structure of CoO as a semiconductor better than GGA-PBE, in agreement with previous studies28 and results reported here are from LDA calculations only. In order to account for the on-site repulsion between d electrons, DFT+U within Dudarev's approach was adopted with U–J parameter values of 4 and 7 for Co and Ni, respectively.29 Wave functions were expanded on a plane wave basis set with a 512 eV kinetic energy cutoff and, in the results for the electronic density of states presented here, a 6 × 6 × 6 k-point BZ sampling was employed. The ground state was in the antiferromagnetic (AFM) configuration. Simulation cells of several sizes were used (with a minimum of four atoms) in order to examine doping in bulk CoO with different Ni concentrations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Special Account for Research Funds of the University of Crete (SARF UOC) (KA 10138).Notes and references
- P. Miretzky and A. F. Cirelli, J. Hazard. Mater., 2010, 180, 1 CrossRef CAS PubMed.
- L. Levankumar, V. Muthukumaran and M. B. Gobinath, J. Hazard. Mater., 2009, 161, 709 CrossRef CAS PubMed.
- D. Wang, Y. Ye, H. Liu, H. Ma and W. Zhang, Chemosphere, 2018, 193, 42 CrossRef CAS PubMed.
- (a) C. Mondal, M. Ganguly, J. Pal, A. Roy, J. Jana and T. Pal, Langmuir, 2014, 30, 4157 CrossRef CAS PubMed; (b) X. Liu, L. Pan, T. Lv, G. Zhu, Z. Suna and C. Sun, Chem. Commun., 2011, 47, 11984 RSC; (c) L. Yang, Y. Xiao, S. Liu, Y. Li, Q. Cai, S. Luo and G. Zeng, Appl. Catal., B, 2010, 94, 142 CrossRef CAS; (d) J. Yu, S. Zhuang, X. Xu, W. Zhu, B. Feng and J. Hu, J. Mater. Chem. A, 2012, 3, 1199 RSC; (e) G. Odling, A. Ivaturi, E. Chatzisymeon and N. Robertson, ChemCatChem, 2017, 10, 234 CrossRef; (f) C. Yu, P. Yang, L. Tie, S. Yang, S. Dong, J. Sun and J. Sun, Appl. Surf. Sci., 2018, 455, 8 CrossRef CAS.
- (a) Z.-J. Jiang and Z. Jiang, Sci. Rep., 2016, 6, 27081 CrossRef CAS PubMed; (b) K. M. Nam, W. S. Seo, H. Song and J. T. Park, NPG Asia Mater., 2017, 9, e364 CrossRef CAS.
- G. Velegraki, J. Miao, C. Drivas, B. Liu, S. Kennou and G. S. Armatas, Appl. Catal., B, 2018, 221, 635 CrossRef CAS.
- (a) D. Gu, C.-J. Jia, C. Weidenthaler, H.-J. Bongard, B. Spliethoff, W. Schmidt and F. Schüeth, J. Am. Chem. Soc., 2015, 137, 11418 Search PubMed; (b) Z. Mao, J. Chen, Y. Yang, D. Wang, L. Bie and B. D. Fahlman, ACS Appl. Mater. Interfaces, 2017, 9, 12427 CrossRef CAS PubMed; (c) L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. R. Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2014, 9, 69 CrossRef CAS PubMed.
- (a) I. T. Papadas, I. Vamvasakis, I. Tamiolakis and G. S. Armatas, Chem. Mater., 2016, 28, 2886 CrossRef CAS; (b) I. T. Papadas, S. Fountoulaki, I. N. Lykakis and G. S. Armatas, Chem. – Eur. J., 2016, 22, 4600 CrossRef CAS PubMed.
- F. Rouquerol, J. Rouquerol and K. S. W. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Academic Press, London, 1999 Search PubMed.
- (a) S. Wang, M. Chen, Y. Xie, Y. Fan, D. Wang, J. J. Jiang, Y. Li, H. Grutzmacher and C. Y. Su, Small, 2016, 12, 2365 CrossRef CAS PubMed; (b) J. S. Gwag and Y. Sohn, Bull. Korean Chem. Soc., 2012, 33, 505 CrossRef CAS.
- J. F. Marco, J. R. Gancedo, M. Gracia, J. L. Gautier, E. I. Rios, H. M. Palmer, C. Greaves and F. J. Berry, J. Mater. Chem., 2001, 11, 3087 RSC.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- H.-K. Lin, H.-C. Chiu, H.-C. Tsai, S.-H. Chien and C.-B. Wang, Catal. Lett., 2003, 88, 169 CrossRef CAS.
- A. Tanaka, K. Nakanishi, R. Hamada, K. Hashimoto and H. Kominami, ACS Catal., 2013, 3, 1886 CrossRef CAS.
- N. Nasrallah, M. Kebir, Z. Koudri and M. Trari, J. Hazard. Mater., 2011, 185, 1398 CrossRef CAS PubMed.
- R. Gherbia, N. Nasrallaha, A. Amraneb, R. Maachia and M. Trari, J. Hazard. Mater., 2011, 186, 1124 CrossRef PubMed.
- A. Zhitkovich, Chem. Res. Toxicol., 2005, 18, 3 Search PubMed.
- (a) Z. B. Alfassi, R. E. Huie, S. Marguet, E. Natarajan and P. Neta, Int. J. Chem. Kinet., 1995, 27, 181 CrossRef CAS; (b) J. Colucci, V. Montalvo, R. Hernandez and C. Poullet, Electrochim. Acta, 1999, 44, 2507 CrossRef CAS; (c) A.-M. Manke, K. Geisel, A. Fetzer and P. Kurz, Phys. Chem. Chem. Phys., 2014, 16, 12029 RSC.
- (a) J. Bisquert, H. Randriamahazaka and G. Garcia-Belmonte, Electrochim. Acta, 2005, 51, 627 CrossRef CAS; (b) L. M. Da Silva, K. C. Fernandes, L. A. De Faria and J. F. Boodts, Electrochim. Acta, 2004, 49, 4893 CrossRef CAS.
- W. S. Seo, J. H. Shim, S. J. Oh, E. K. Lee, N. H. Hur and J. T. Park, J. Am. Chem. Soc., 2005, 127, 6188 CrossRef CAS PubMed.
- http://www.metrohm.com/en/applications/# .
- P. I. Ravikovitch, D. Wei, W. T. Chueh, G. L. Haller and A. V. Neimark, J. Phys. Chem. B, 1997, 101, 3671 CrossRef CAS.
- P. Kubelka, J. Opt. Soc. Am., 1948, 38, 448 CrossRef CAS PubMed.
- (a) G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS; (b) G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS; (c) G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- (a) P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef; (b) P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223 CrossRef; (c) G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- N. Alidoust, M. Lessio and E. A. Carter, J. Appl. Phys., 2016, 119, 025102 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical data, TGA profile, TEM images, XRD patterns, N2 physisorption isotherms, XPS spectra, FL spectra and catalytic data of Co1−xNixO MNAs. See DOI: 10.1039/c8qi01324a |
| This journal is © the Partner Organisations 2019 |