
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular complexation between chain-folding poly(ester-imide)s and polycyclic aromatics: a fractal-based pattern of NMR ring-current shielding†
Marcus
Knappert
a,
Tianqi
Jin
a,
Scott D.
Midgley
a,
Guanglu
Wu
 b,
Oren A.
Scherman
b,
Ricardo
Grau-Crespo
a and
Howard M.
Colquhoun
*a
b,
Oren A.
Scherman
b,
Ricardo
Grau-Crespo
a and
Howard M.
Colquhoun
*a
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: h.m.colquhoun@rdg.ac.uk
bMelville Laboratory for Polymer Synthesis, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
First published on 13th November 2019
Abstract
Polycondensation of the diimide-based diols N,N′-bis(2-hydroxyethyl)hexafluoro-isopropylidene-diphthalimide (HFDI), N,N′-bis(2-hydroxyethyl)pyromellitimide (PMDI), or N,N′-bis(2-hydroxyethyl)naphthalene-1,4,5,8-tetracarboxylic-diimide (NDI) with aliphatic diacyl chlorides ClOC(CH2)xCOCl (x = 1 to 8) affords linear poly(ester-imide)s. Homopolymers based on HFDI do not interact with polycyclic aromatics such as pyrene and perylene, as demonstrated by 1H NMR spectroscopy. However, poly(ester-imide)s containing NDI residues show significant upfield complexation shifts of the diimide resonance in the presence of pyrene and perylene, consistent with supramolecular binding of the polycyclic aromatic molecules at the diimide residues. The latter series of poly(ester-imide)s (x = 1 to 8) shows a maximum in complexation shift of the NDI resonance at x = 2. Computational simulations using a density functional based tight-binding (DFTB) method suggest that the maximum at x = 2 is due to the presence of chain folds that are geometrically optimum for a pyrene molecule to bind between pairs of adjacent NDI residues, making near-van-der-Waals contact with both diimide units. As a test of this binding model, 1H NMR studies of pyrene complexation with a copoly(ester-imide) containing both NDI and HFDI units (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio, x = 2) were carried out. The resulting NDI resonance-pattern showed clear evidence of fractal-type character and confirmed tight chain-folding and pairwise binding of pyrene.
:1 mole ratio, x = 2) were carried out. The resulting NDI resonance-pattern showed clear evidence of fractal-type character and confirmed tight chain-folding and pairwise binding of pyrene.
1. Introduction
The ability of linear copolymers such as DNA and RNA to store and process digital sequence-information underpins the whole of biology.1,2 However, no comparable information technology has yet been developed for molecular systems based on synthetic copolymers, even though the simplest AB copolymer is the logical equivalent of a binary string.3 Nevertheless, significant progress towards such technology has been made in recent years,4 especially with the development of sequence-specific polymerisation (i.e. sequence-“writing”) techniques.5–7 In terms of “reading” copolymer sequences, it has been shown that tweezer-type reporter molecules with pyrene-based, π-electron-rich arms bind to copolymers containing π-electron-poor pyromellitimide (PMDI) or naphthalene-1,4,5,8-tetracarboximide (NDI) residues, via complementary π–π-stacking.8–10 This type of binding results in upfield shifts and splittings of 1H NMR resonances associated with the diimide residues, as a result of ring-current shielding by the complexed aromatic rings. This effect has also been discussed in other, related work on polymer chain-folding induced by π–π-stacking.11–14 It has further been shown that additive, long range ring-current shielding by tweezer molecules bound further out along the chain increases the complexation shifts of NDI resonances, resulting in these shifts being highly sequence-dependent.9 In addition, different tweezer-molecules were found to bind to different triplet-sequences, thereby producing a “frameshift” effect in the binding of longer sequences by multiple tweezer-molecules.10More recently, it has been found that even simple non-tweezer molecules such as pyrene and perylene can produce sequence-dependent 1H NMR complexation shifts in certain copolyimides where tight chain-folding enables the small molecule to bind via intercalation between two adjacent NDI residues.15 Curiously, the resulting pattern of 1H NMR resonances shows a marked degree of self-similarity, and it was found that this reflects an underlying fractal distribution of ring-current shieldings produced by pyrene molecules binding to all the allowed sequences within which an NDI residue may be embedded.
In a new investigation we have studied the binding of π-electron-rich aromatic molecules, specifically pyrene and perylene, to a novel series of homo- and co-poly(ester-imide)s containing strongly-binding NDI residues, weakly-binding PMDI residues and/or non-binding hexafluoro-isopropylidene-diphthalimide (HFDI) sub-units. The work was undertaken because poly(ester-imide)s16–19 have much greater future potential for “writing” sequence information (via catalytic ester-interchange) than the simpler, more chemically inert poly(ether-imide)s investigated previously.15 In the present paper we report the discovery of an optimum poly(ester-imide) structure for chain-folding and pyrene-binding, identified by systematically varying the diester spacer-unit between adjacent diimide (NDI) residues.
2. Experimental section
2.1 Materials and instrumentation
Starting materials, solvents, diacyl chlorides ClCO(CH2)xCOCl (x = 1–4 and 6–8), and heptanedioic acid were purchased from Sigma-Aldrich, ThermoFisher or Fluorochem and were used as received. The diimide-based diols N,N′-bis(2-hydroxyethyl)hexafluoro-isopropylidene-diphthalimide (1)20N,N′-bis(2-hydroxyethyl)-pyromellitimide (2),21 and N,N′-bis(2-hydroxyethyl) naphthalene-1,4,5,8-tetracarboxylic diimide (3)22 were synthesised as noted below. Proton and 13C NMR spectra were recorded on a Bruker Nanobay 400 spectrometer (400 MHz for 1H and 100 MHz for 13C NMR) and on a Bruker AVANCE 500 spectrometer with TCI Cryoprobe system (500 MHz for 1H NMR). Proton chemical shifts (δ) are reported in ppm relative to tetramethylsilane (TMS, δ = 0.00 ppm), referred to residual 1H solvent peaks, and 13C NMR chemical shifts are similarly reported relative to TMS. Fourier transform infrared (FTIR) spectra were recorded on a Perkin Elmer 100 Spectrum FT-IR using a diamond ATR sampling accessory. Mass spectrometry was carried out using a ThermoFisher Scientific Orbitrap XL LCMS. Gel permeation chromatography (GPC) was conducted using an Agilent Technologies 1260 Infinity system calibrated with polystyrene standards. Samples were dissolved in analytical grade THF (2 mg mL−1) containing BHT as stabiliser and run using the same solvent as the mobile phase, eluting through two Agilent PLgel 5 μm MIXED-D 300 × 7.5 mm columns in series. Phase transitions were identified by DSC under nitrogen, using a TA Instruments DSC 2920 or a Mettler Toledo DSC23e instrument. Melting points are quoted as peak temperatures and glass transitions as onsets. Weighed samples of ca. 5 mg of monomer or 10 mg of polymer were heated from 30 °C to 325 °C, twice, at a scan rate of 10 °C min−1. Inherent viscosities (ηinh) were measured at 25 °C with 0.1% w/v solutions of polymer in chloroform/hexafluoropropan-2-ol (1:1, v/v) or in chloroform/trifluoroethanol (6:1, v/v) using a Schott Instruments CT 52 auto-viscometer with glass capillary no. 53103.
2.2 Computational methods
Pyrene binding (intercalation) energies were obtained using the self-consistent-charge density functional tight-binding (SCC-DFTB) approach, as implemented within the DFTB+ code.23 Parameters for all atoms and pairs including elements C, H, N, O were taken from the “mio” parameter set of the Slater–Koster library.24 Dispersion corrections based on a Lennard-Jones potential were applied in all simulations.25 Each polymer was modelled as a discrete heptameric oligomer, and the intercalation energy of pyrene was evaluated at the central chain-fold. Intercalation energies (Eint) were derived using the expression:| Eint = Ecomplex − (Epyrene + Epolymer) |
Simulations of 1H NMR spectra were carried out using the “peak table to spectrum” script within Mnova (version 14.1, Mestrelab Research, Santiago de Compostela).
2.3 Monomer synthesis
2.4 Polymer synthesis: representative procedure
1,2-Dichlorobenzene (4.5 mL, distilled from CaH2), N,N′-bis-(2-hydroxyethyl)-naphthalene tetracarboxylic diimide (dried at 120 °C for 24 h; 2.09 g, 5.90 mmol) and adipoyl chloride, ClCO(CH2)4COCl (1.09 g, 5.96 mmol), were stirred under nitrogen at room temperature and the mixture was heated to 170 °C for 24 h with a slow dinitrogen purge to remove the evolved hydrogen chloride. After cooling to room temperature the reaction mixture was dissolved in dichloromethane/hexafluoroisopropanol (1:1, v/v, 30 mL) and added dropwise into an excess of methanol (400 mL). The precipitate was filtered off, washed with methanol and dried at 80 °C for 24 h. The reprecipitation was repeated three times to afford pure polymer (2.10 g, 76%). Variations from this procedure for specific polymers are noted in Table 1, and full synthetic details are given in the ESI.†
| Polymer ref. no. | Diimide monomer |
x in (CH2)xd |
Reaction solvent | Temp. (°C) | Time (h) | η inh (dL g−1) | M n (daltons) | Đ | T g (°C) | T m (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Full details in ESI.† N,N′-Bis-(2-hydroxyethyl)hexafluoroisopropylidene diphthalimide. b N,N′-Bis-(2-hydroxyethyl)pyromellitic diimide. c N,N′-Bis-(2-hydroxyethyl)-1,4,5,8-naphthalenediimide. d In diacid chloride ClOC(CH2)xCOCl. e 1,2-Dichlorobenzene. f 1-Chloronaphthalene. | ||||||||||
| 4 | HFDIa | 3 | 1,2-DCBe | 120 | 4 | 0.83 | 30200 |
2.07 | 109 | — |
| 5 | HFDI | 5 | 1-CNpf | 120 | 4 | 0.56 | 20400 |
1.92 | 72 | — |
| 6 | PMDIb | 1 | 1,2-DCB | 170 | 24 | 0.48 | — | — | — | 194 |
| 7 | PMDI | 2 | 1,2-DCB | 170 | 24 | 0.36 | — | — | — | 233 |
| 8 | PMDI | 3 | 1,2-DCB | 170 | 24 | 0.60 | — | — | — | 223 |
| 9 | PMDI | 4 | 1,2-DCB | 170 | 24 | 0.55 | — | — | — | 253 |
| 10 | PMDI | 5 | 1,2-DCB | 170 | 24 | 0.59 | — | — | — | 190 |
| 11 | PMDI | 6 | 1,2-DCB | 170 | 24 | 0.62 | — | — | — | 217 |
| 12 | PMDI | 7 | 1,2-DCB | 170 | 24 | 0.37 | — | — | — | 203 |
| 13 | PMDI | 8 | 1,2-DCB | 170 | 24 | 0.59 | — | — | — | 207 |
| 14 | NDIc | 1 | 1,2-DCB | 170 | 24 | 0.17 | — | — | 189 | — |
| 15 | NDI | 2 | 1,2-DCB | 170 | 24 | 0.56 | — | — | 139 | — |
| 16 | NDI | 3 | 1,2-DCB | 170 | 24 | 1.54 | — | — | 132 | — |
| 17 | NDI | 4 | 1,2-DCB | 170 | 24 | 0.75 | — | — | 116 | — |
| 18 | NDI | 5 | 1,2-DCB | 170 | 24 | 0.58 | — | — | 90 | — |
| 19 | NDI | 6 | 1,2-DCB | 170 | 24 | 0.19 | — | — | 73 | — |
| 20 | NDI | 7 | 1,2-DCB | 170 | 24 | 1.20 | — | — | 76 | — |
| 21 | NDI | 8 | 1,2-DCB | 170 | 24 | 0.93 | — | — | 50 | — |
| 22 | NDI/HFDI | 2 | 1-CNp | 160 | 24 | 0.47 | — | — | 130 | — |
| 23 | NDI/HFDI | 3 | 1-CNp | 160 | 24 | 0.26 | — | — | 99 | — |
2.5 Polymer characterisation
Poly(ester-imide)s 4–25 were characterised by 1H and 13C NMR spectroscopy, IR spectroscopy, DSC, and solution viscometry. Results are summarised in Table 1 and detailed in the ESI.† Gel permation chromatography in THF was possible for homopolymers 4 and 5, derived from 4,4′-hexafluoroisopropylidenediphthalic dianhydride, but all the other polymers described here were insoluble in conventional GPC solvents and so were characterised by solution viscometry in the proton-donor solvent system chloroform/trifluoroethanol (6:1, v:v). However, the GPC data for 4 and 5 enabled construction of a calibration plot of inherent viscosity against Mn (see ESI†) and, from this plot, estimates of Mn for the poly(ester-imide)s reported here could be obtained if required.
3. Results and discussion
3.1 Synthesis of poly(ester-imide)s
The aim of the present work was to evaluate the possibility of supramolecular complexation of small molecules with poly(ester-imide)s via chain-folding and complementary π–π-stacking. We have shown previously that supramolecular processes of this type lead to sequence-dependent complexation shifts of the diimide 1H NMR resonances, which thus enable the “reading” of sequence-information in poly(ether-sulfone-imide)s. This has been demonstrated using not only tweezer-type molecules8–10 but also simple polycyclic aromatics such as pyrene.15 To identify the optimum poly(ester-imide) structure for such binding, homopolymers were synthesised by condensation of the diimide-based diols 1–3 (Fig. 1) with the diacyl chlorides ClOC(CH2)xCOCl (x = 1 to 8). Reactions were carried out at high temperature (120 to 170 °C) in 1,2-dichlorobenzene or 1-chloronaphthalene under a slow dinitrogen purge, and the resulting polymers (Table 1 and ESI†) were isolated and purified by reprecipitation from mixed proton-donor solvents such as chloroform/trifluoroethanol or dichloromethane/hexafluoroisopropanol. Inherent viscosity (ηinh) and GPC data for polymers derived from diol 1 (the only THF-soluble polymers obtained in this work) indicated that viscosities of ca. 0.5–0.8 dL g−1 corresponded to molecular weights (Mn) in the range 20000–30000 daltons, with dispersities very close to the theoretical step-growth value of two.
 | ||
| Fig. 1 Imide-diol monomers, poly(ester-imide)s or copoly(ester-imide)s, with reference numbers. | ||
3.2 Interactions of poly(ester-imide)s with polycyclic aromatics
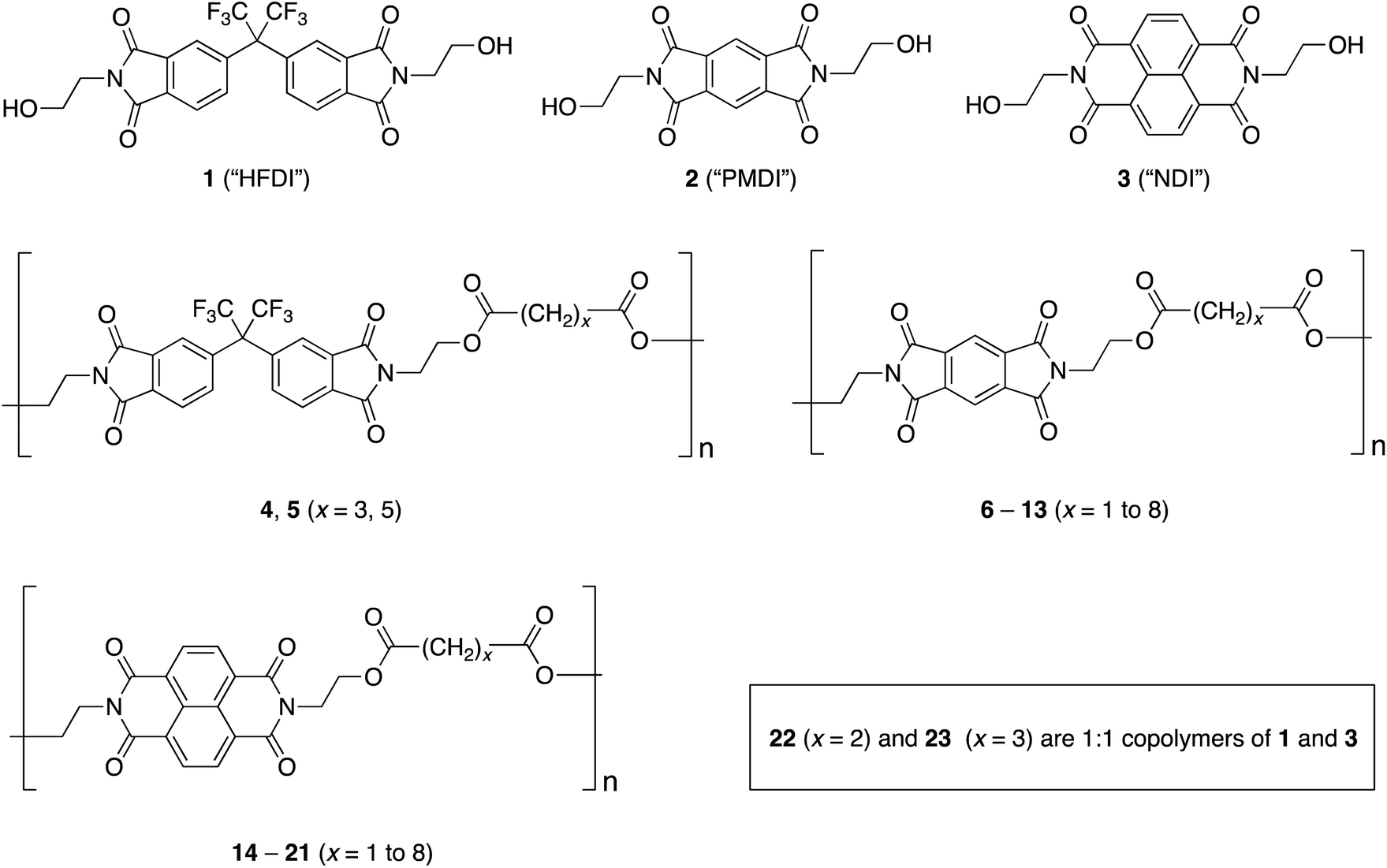
It is well established that complexation of certain polyimides by electron-rich aromatic units such as pyrene and perylene, via complementary π–π-stacking, leads to substantial upfield complexation shifts of 1H NMR resonances associated with the diimide residues, as a result of magnetic ring-current shielding by the complexed aromatic unit.8–15 Although the forces involved in complementary π–π-stacking remain a matter for discussion, it is clear that this type of supramolecular complexation is favoured by coplanarity of the diimide residues, as in PMDI and NDI, and strongly disfavoured by non-planar and/or sterically-hindered diimide units such as HFDI.Thus, the HFDI-based poly(ester-imide)s 4 and 5 (4 mM solutions based on diimide residues) showed no detectable 1H NMR complexation shifts in the presence of pyrene or perylene (2 equivalents per diimide residue), whereas the PMDI-based polymers 6 to 13 showed small but consistent upfield shifts (ca. 0.1 ppm) of their PMDI resonances under the same conditions. A very slightly enhanced complexation shift was observed at x = 2, but otherwise there was no significant dependence of the complexation shift on the length of the diester-linkage between adjacent PMDI residues (see ESI†). In contrast, the NDI resonances in polymers 14 to 21 showed substantial complexation shifts in the presence of 2 equivalents of pyrene, the shifts varying markedly (from 0.20 to 0.75 ppm at 8 mM concentration of NDI residues) with the length of the diester-spacer [–OOC(CH2)xCOO–]. Polymer-pyrene binding evidently occurs under fast-exchange conditions on the NMR time scale because separate resonances corresponding to bound and unbound NDI residues are not observed, even at sub-stoichiometric pyrene:NDI ratios. The NDI resonance-positions represent time-averaged chemical shifts for bound and unbound residues. They thus move progressively to higher field (to a greater or lesser extent, depending on the sequence) as increasing pyrene concentration shifts the equilibrium towards the bound state. The complexation shift reached a maximum at x = 2, and the same trend was found for complexation of perylene, which produced even more pronounced complexation shifts (Fig. 2). Charge-transfer absorptions resulting from polyimide complexation with these aromatic molecules produce strongly coloured solutions – deep red for pyrene and an intense dark green for perylene.11
 | ||
| Fig. 2 Complexation shifts for NDI-based poly(ester-imide)s 14–21 [8 mM in NDI, in CDCl3/TFE (6:1 v/v)] in the presence of 2 mol. equiv. per NDI of pyrene-d10 (△) or perylene-d12 (□). | ||
3.3 Computational simulation of pyrene-binding
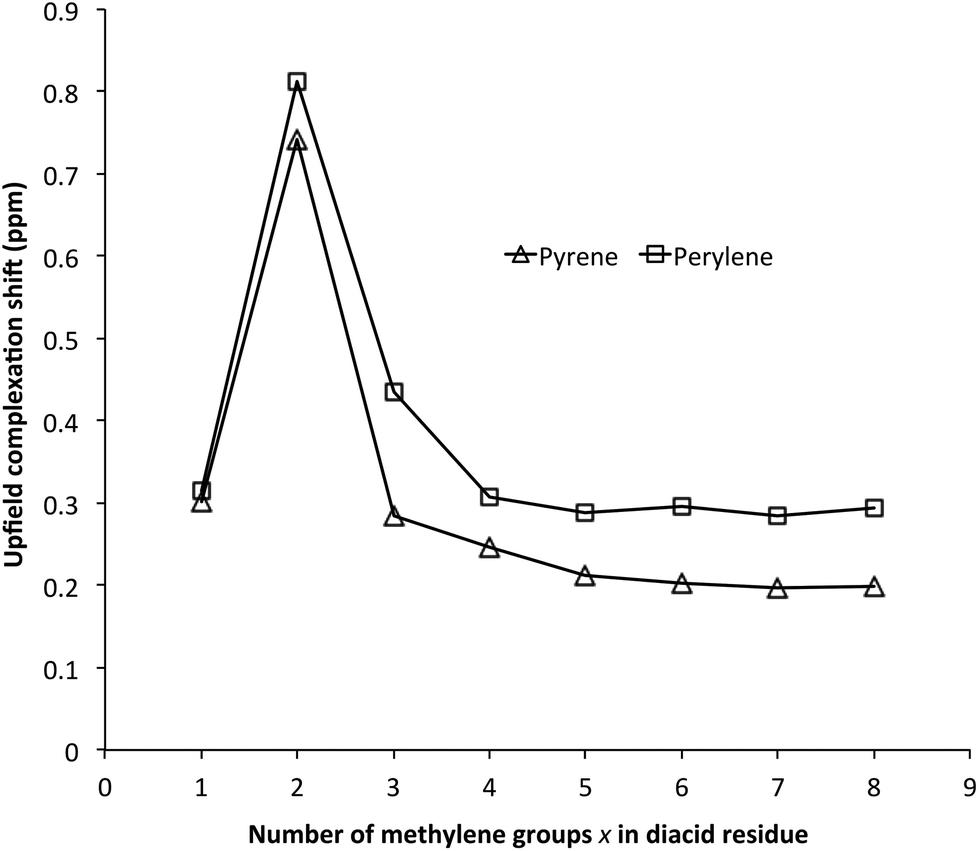
In light of previous work showing that strong complexation of pyrene by poly(ether-imide)s was associated with chain-folding and binding of the small molecule by intercalation between two neighbouring NDI residues,15 it seemed likely that the maximum in pyrene binding at x = 2 results from the polymer with this spacer adopting a chain-folded geometry that is especially favourable for binding. Computational simulation (SCC-DFTB with dispersion correction) of pyrene complexation by polymers 14 (x = 1) 15 (x = 2) and 18 (x = 5) provided strong support for this idea, with computed binding energies for pyrene indeed being greatest at x = 2, as shown in Fig. 3. The polymers were modelled as heptamers, with the energies of binding being calculated for a pyrene molecule intercalating at the central chain-fold.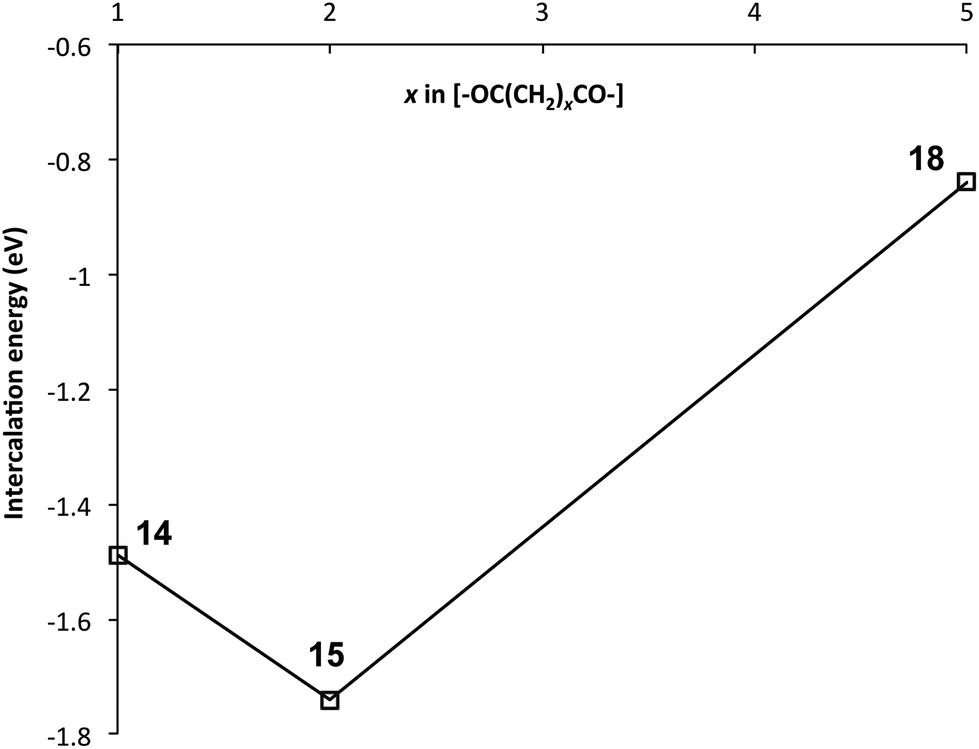 | ||
| Fig. 3 Computed intercalation energies for the binding of pyrene to poly(ester-imide)s 14 (x = 1) 15 (x = 2) and 18 (x = 5). More negative energy-values correspond to stronger binding. | ||
A rationale for these results can be seen immediately by inspection of Fig. 4, which shows space-filling (van der Waals radii) models of energy-minimised chains for the three different values of x, in the presence of an intercalating pyrene molecule. For x = 1 the chain is seen to fold regularly, with near-parallel NDI subunits, but the average C–C distance between equivalent NDI carbons at the “binding” chain fold of 8.14 Å (range 8.00 to 8.27 Å) is clearly too great for pyrene to make simultaneous van der Waals contact with both NDI residues. For x = 2, however, the chain-fold is tighter and the gap between neighbouring NDI residues is smaller (average 7.59 Å), even though the diester spacer is longer and the binding residues are tilted slightly towards one other (contact range 6.67 to 8.50 Å). This tighter chain-folding for x = 2 leads to the heptameric chain becoming noticeably contracted relative to the corresponding chain where x = 1 (Fig. 4), and the intercalating pyrene makes much closer contact with both adjacent NDI residues. Finally, when x = 5, the chain folding is distinctly irregular, and the NDI-NDI spacing at the “binding” chain fold is greatly expanded to give an average C–C spacing of 10.02 Å (range 9.80 to 10.24 Å). The trend in binding strength for polymers 14, 15 and 18, where x = 1, 2 and 5 respectively, is thus reasonably accounted for.
 | ||
| Fig. 4 Energy-minimised geometries (SCC-DFTB with dispersion correction) for pyrene complexes of the NDI-based poly(ester-imide)s 14, 15 and 18, modelled as heptamers. The optimum fit for π–π-stacking of pyrene at x = 2 (polymer 15) is very clear. | ||
3.4 Sequence-dependent binding to a co-poly(ester-imide)
Our earlier work on sequence-dependent polymer complexation with pyrene showed that an NDI copolyether containing equal proportions of strongly-binding and weakly-binding chain folds (Fig. 5a/b) gave, in the presence of pyrene, a 1H NMR spectrum containing a characteristic set of NDI resonances, with each resonance corresponding to a specific NDI-centred sequence or group of sequences.15 This resonance pattern was shown to result from intercalation of pyrene at tight chain-folds formed by pairs of NDI residues linked by triethylene-dioxy spacer units (Fig. 5a), and the pattern could in fact be accounted for on the basis that binding at any other NDI residue in the chain was negligible by comparison. Moreover, the NMR spectrum itself was seen to have elements of self-similarity, arising from an underlying fractal distribution of ring-current shieldings produced by pyrene-intercalation at all possible NDI-centred sequences.15 In the present poly(ester-imide)s, an analogous situation could exist for the 1:1 copolymer containing bis(oxyethyl)-NDI and -HFDI residues linked by the succinate spacer (x = 2, copolymer 22), as shown in Fig. 5d. As a test of the tight chain-folding model, we therefore synthesised copolymer 22 and carried out a 1H NMR titration against pyrene-d10 (see ESI†). The theory underlying this test is outlined below for quintet sequences, and is described in more detail for septet and nonet sequences in ref. 15.
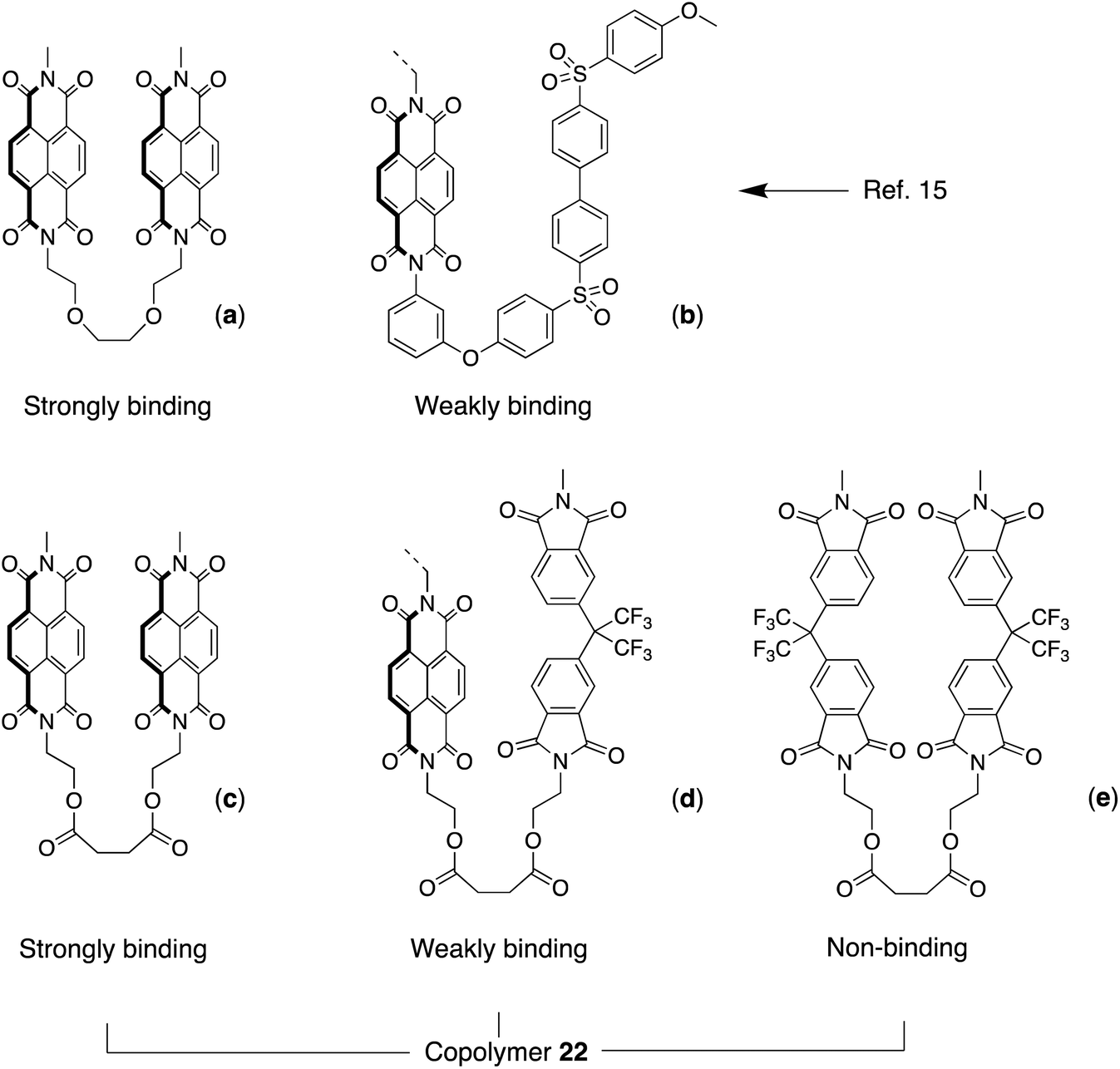 | ||
| Fig. 5 (a) Possible chain folds in: (a)/(b) the copoly(ether-imide) described in ref. 15; and (c)/(d)/(e) copoly(ester-imide) 22 (this work). | ||
On the basis that, in copolymer 22, we can identify a pair of adjacent NDI residues as a strongly-binding site, and ignore any weak binding at other positions on the chain, it is possible to assign a digital code to each NDI-centred sequence. This code describes the location and number of such “pairwise” binding sites relative to the central NDI residue. For example, denoting NDI residues as “I” and HFDI residues as “F”, the quintet sequence IFIII has one “II” pair adjacent to (and including) the central “I” and one pair at a next-adjacent position. It is therefore assigned the code 11. The sequence FIIIF, in contrast, has two “II” pairs adjacent to the centre, and zero pairs at the next-adjacent position, so this sequence would be assigned the code 20. Table 2 shows all sixteen possible I-centred quintet sequences and their assigned two-digit codes. There are nine possible combinations of 0, 1 and 2, taken in twos, but three of these (01, 02 and 12) are unused because they do not represent any of the sixteen possible quintet sequences. The table also shows that four of the six assigned codes are degenerate, i.e. they represent more than one sequence: much of this degeneracy arises from the fact that mirror-image copolymer sequences (shown side-by-side in Table 2) are indistinguishable by NMR.
 ) are total shielding factors for the originating sequences, calculated from eqn (1) (below) with the value of a set to unity
) are total shielding factors for the originating sequences, calculated from eqn (1) (below) with the value of a set to unity

|
The significance of these digital codes is that, as described in an earlier paper,15 they can predict the total ring-current shielding by an intercalating aromatic molecule of a central “observed” NDI residue in any given sequence. Such shielding, is additive, with an empirical “fall-off factor” of about 4 as an aromatic molecule such as pyrene binds at chain-folds further and further out from the central diimide residue. The system is digital rather than analogue because there are no intermediate binding positions between NDI residues, and also because there are just three possibilities (0, 1 or 2) for the number of pyrene molecules bound at any specific distance (viewed in both directions) from the central NDI residue.
Using the above concepts, it may be shown that the total (relative) ring current shielding, T, of a central NDI residue by pyrene molecules intercalating into a specific binary copolymer sequence can be described quantitatively by eqn (1):15
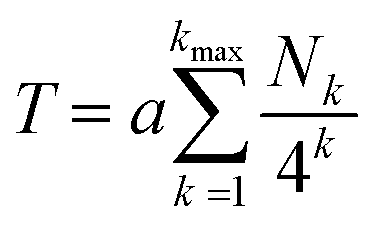 | (1) |
3.5 Fractal character of ring-current shieldings in copolymer 22
We have shown previously15 that eqn (1) (with a = 1 and kmax = ∞) defines a mathematical fractal known as the fourth-quarter Cantor set (even though this set was originally discovered by Smith).26 The set represented by eqn (1) can, like many fractals, also be constructed geometrically. In the present case, a line of unit length is divided into four equal segments and the right hand (fourth) segment is discarded. These two operations are then repeated on the remaining three segments, and so on indefinitely (see Fig. 6). In copolymer terms, successive iterations of this construction correspond to increasing the length of the copolymer sequence being considered. Thus, the first iteration yields three segments corresponding – in a copolymer containing only NDI (“I”) and HFDI (“F”) residues – to the three distinguishable triplet sequences FIF, [FII + IIF] and III. The latter sequences contain 0, 1 and 2 “II” pairs, respectively, and the corresponding segments are thus labelled 0, 1 and 2. The second iteration of the construction yields nine segments labelled 00, 01, 02 etc., corresponding to the nine possible combinations of 0, 1 and 2 taken in pairs, as shown in Table 2. However, Table 2 also demonstrates that only six of these nine combinations are digital codes corresponding to actual and distinguishable F/I quintets, so just six quintet-based resonances are predicted by the model (Fig. 6). Further iterations of the construction would yield many more predicted resonances, corresponding to longer and longer sequences, but only quintet sequences are resolved in the present experimental study (Fig. 7 and 8).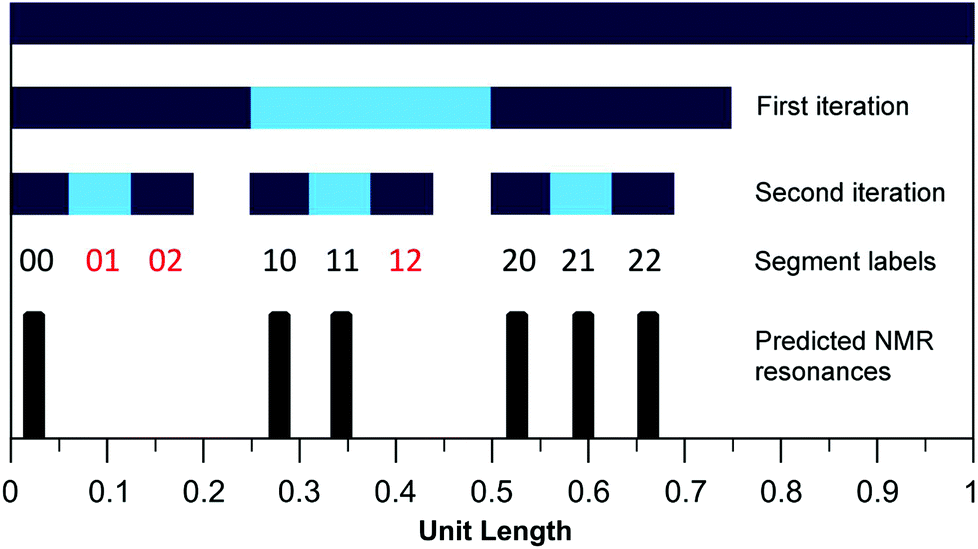 | ||
Fig. 6 Graphical construction of the fourth-quarter Cantor set (first two iterations). This shows how the relative positions of the 1H NMR resonances arising from the quintet sequences shown in Table 2 emerge from the two-digit codes assigned to each sequence using a chain-folded “pairwise” binding model for pyrene. The assignment process does not generate the codes 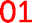 , ,  and and  (Table 2), so it is predicted that no corresponding NMR resonances will be found at these positions. (Table 2), so it is predicted that no corresponding NMR resonances will be found at these positions. | ||
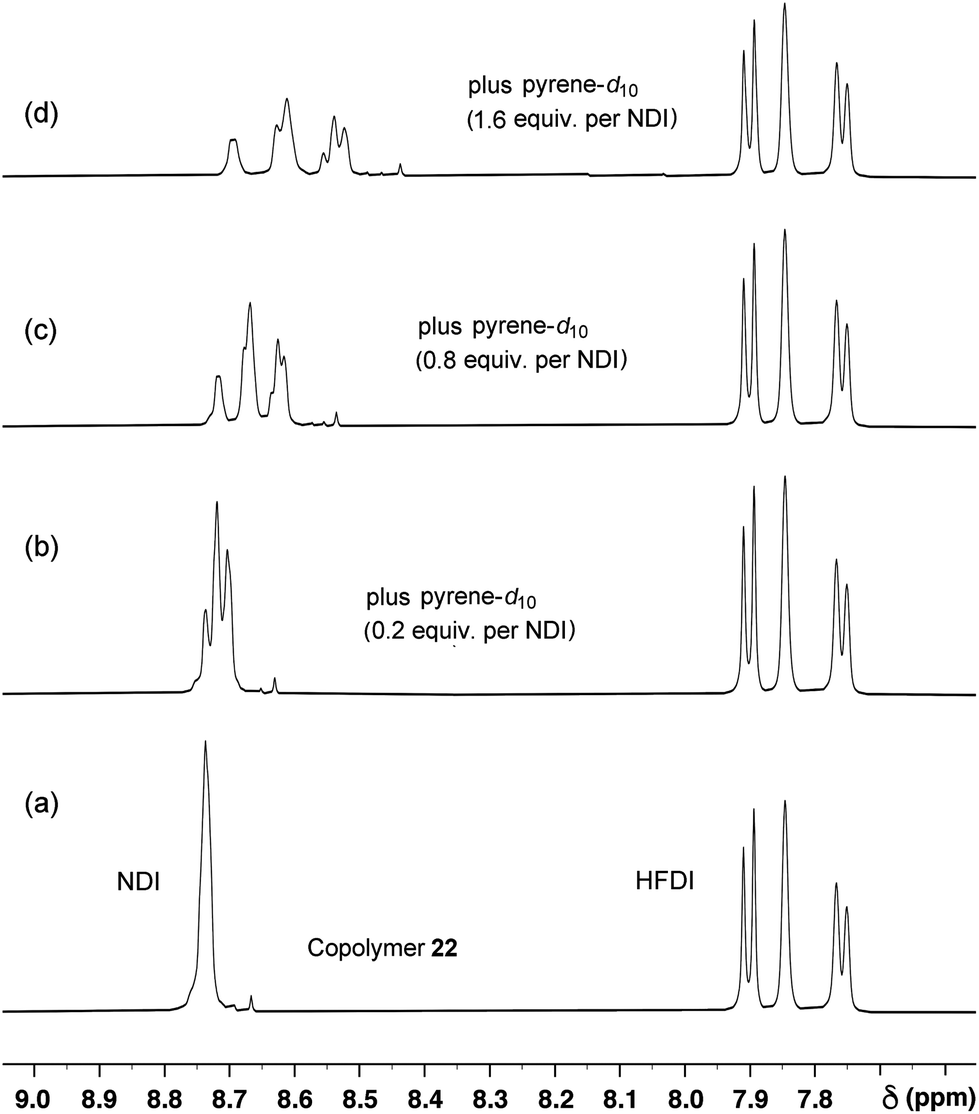 | ||
| Fig. 7
1H NMR titration of copolymer 22 in CDCl3/trifluoroethanol (6:1 v/v) against pyrene-d10. As the concentration of pyrene increases, the single NDI (“I”) resonance in the absence of pyrene splits into three signals corresponding to the three distinguishable triplet sequences IFI, [IIF + FFI], III. Ultimately, six NDI resonances are seen corresponding to the six groups of quintets shown in Table 2. The very weak resonance at ca. 8.67 ppm in the spectrum of the uncomplexed polymer is assigned to a trace of NDI-based macrocycle (probably the cyclic dimer shown in the ESI†), as it shows a strong upfield shift in the presence of pyrene but no sequence-related splitting. | ||
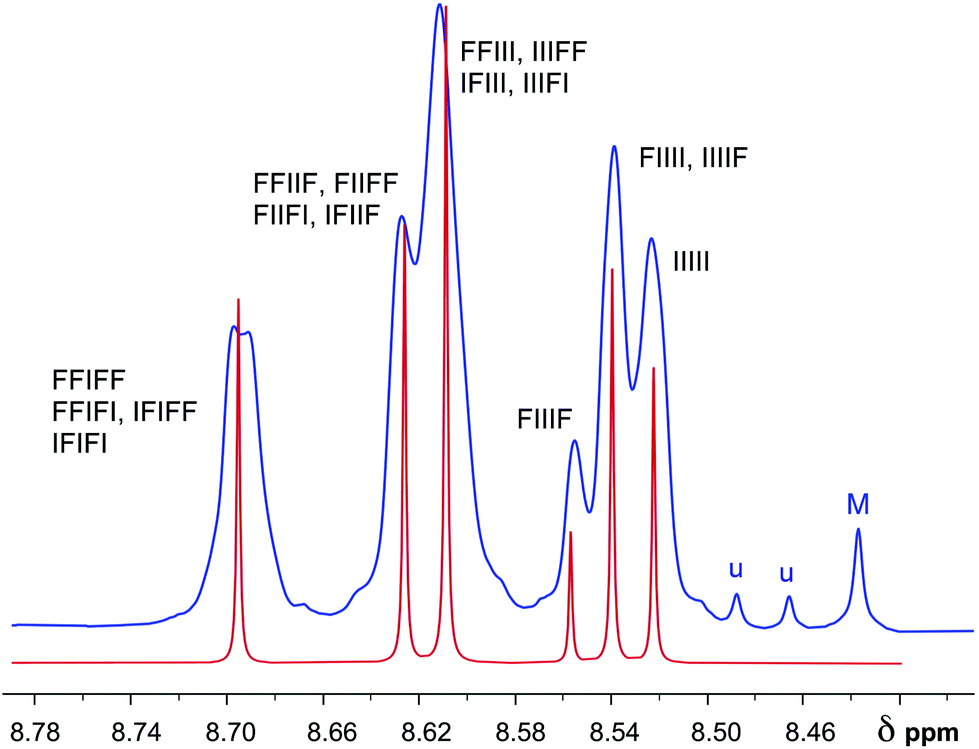 | ||
| Fig. 8 Comparison of simulated (red) and experimental (blue) 1H NMR resonance patterns (NDI protons only) for copolymer 22 in the presence of 1.6 mole equivalents of pyrene per NDI residue. The simulated spectrum was generated using the total shielding parameters T (Table 2) for all possible NDI/HFDI quintet sequences, scaled empirically by a value for a (eqn (1)) of 0.368 to afford complexation shifts in ppm. Relative intensities were simulated as sequence-probabilities, calculated on the basis of a copolymer chain in which the sequence-distribution is 80% random relative to a non-random diblock NDI/HFDI copolymer. (M = probable macrocycle; u = unidentified). | ||
Indeed, titration of copolymer 22 against pyrene-d10 (Fig. 7) resulted in the emergence, above a pyrene:NDI-residue mole ratio of ca. 1:1, of a resonance-pattern [spectrum 7(b), mole ratio 1.6:1] whose relative chemical shifts in the NDI region correspond very closely to those predicted in Fig. 6. This result provides strong support for our proposal (Fig. 4) that the observed peak in binding strength for the diester linker with x = 2 is associated with tight chain-folding and “pairwise” binding of each pyrene to two adjacent NDI residues.
Prediction of an NMR spectrum requires not only chemical shift values but also resonance intensities. For signals arising from different copolymer sequences, such intensities are directly proportional to sequence-probabilities, but in a 100% random, 1:1 copolymer all sequences of a given length would have the same probability. Different resonance intensities would then simply reflect the number of different sequences contributing to each resonance.
For copolymer 22, the observed resonance pattern [spectrum 7(d)] corresponds, as shown below, to a copolymer sequence-distribution having a degree of randomness of ca. 80%. In a fully random I/F copolymer (1:1 mole ratio) the probability of finding “F” or “I” at any (non-centred) position in an NDI-centred sequence is 0.5, but at 80% randomness (relative to a 1:1 diblock [(NDI)n–(HFDI)n] copolymer defining a randomness of zero) the probability of finding “F” at any position in an NDI-centred sequence would be 0.4, and that of finding “I” 0.6. Since copolymer 22 has overall 1:1 stoichiometry – corresponding to the monomer feed-ratio and also confirmed by integration of NMR spectrum 7(a) – this also implies that the HFDI-centred sequences must contain “F” with probability 0.6 and “I” with probability 0.4. In other words, the copolymer exhibits some degree of blockiness. The methodology for simulating 1H NMR spectra at varying degrees of chain-randomness is detailed in the ESI.†
On this basis it proved possible to simulate a 1H NMR spectrum for the NDI resonances of copolymer 22 in the presence of pyrene, using complexation shifts (Table 2) predicted by eqn (1) and quintet sequence-probabilities calculated on the basis of an 80% degree of randomness. The simulated spectrum is shown in Fig. 8, superimposed on the experimental spectrum [an expanded version of spectrum 7(d)]. The agreement is not quite perfect (a very weak splitting of the lowest-field resonance is, for example, unaccounted for) but it is certainly good enough to confirm the hypothesis that the peak in binding energy for pyrene at x = 2 results from tight chain-folding and pairwise binding at adjacent NDI residues. The weak NDI resonance at highest field (ca. 8.44 ppm in Fig. 8) marked “M” is assigned to trace levels of an NDI-based macrocycle, most probably a dimer (see ESI†) as this resonance also shifts markedly to high field in the presence of pyrene but shows no sequence-related splitting. Macrocycles based on NDI are of course very well-known,27,28 and indeed one such pyrene-binding macrocycle has previously been isolated and identified from the synthesis of a related poly(ether-imide).29
4. Conclusions
Poly(ester-imide)s obtained by polycondensation of N,N′-bis(2-hydroxyethyl)naphthalene-1,4,5,8-tetracarboxylic-diimide with the aliphatic diacyl chlorides ClOC(CH2)xCOCl (x = 1 to 8) show significant upfield complexation shifts of the diimide resonance in the presence of pyrene and perylene, as a result of supramolecular binding of the polycyclic aromatic molecules at the diimide residues. Pronounced maxima in the complexation shifts with these hydrocarbons are seen for the poly(ester-imide) with x = 2, due to the presence of a chain fold that is geometrically optimum for an aromatic molecule to intercalate into and make near-van-der-Waals contact with two ajacent diimide residues. “Pairwise” binding of this type is confirmed, both computationally and by 1H NMR studies of pyrene complexation with a 1:1 copoly(ester-imide), again with x = 2, but now containing both NDI and HFDI units. The resulting NMR resonance-pattern shows clear evidence of fractal-type character and this pattern is shown to be fully consistent with previous results for tight chain-folding and “pairwise” binding of pyrene to NDI residues.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was sponsored by the H2020 program of the European Union under the ITN project Euro-Sequences, H2020-MSCA-ITN-2014, grant number 642083 (Marie Skłodowska-Curie PhD studentship to MK). Further support was provided by the Leverhulme Trust, (Emeritus Fellowship to HMC, grant number EM-2018-0161/4, and postdoctoral support to GW – Natural Material Innovation for Sustainable Living); and by EPSRC (programme grant number EP/L027151/1, to OAS).References
- F. H. C. Crick, On protein synthesis, Symp. Soc. Exp. Biol., 1958, 12, 138–163 CAS.
- M. Nirenberg, Historical review: Deciphering the genetic code – a personal account, Trends Biochem. Sci., 2004, 29, 46–54 CrossRef CAS PubMed.
- C. R. Dawkins, The Blind Watchmaker, Longmans, London, 1986, p. 115 Search PubMed.
- M. G. T. A. Rutten, F. W. Vaandrager, J. A. A. W. Elemans and R. J. M. Nolte, Encoding information into polymers, Nat. Rev. Chem., 2018, 2, 365–381 CrossRef.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Sequence-Controlled Polymers, Science, 2013, 341, 1238149 CrossRef PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, From precision polymers to complex materials and systems, Nat. Rev. Chem., 2016, 1, 1–14 Search PubMed.
- J.-F. Lutz, Defining the field of sequence-controlled polymers, Macromol. Rapid Commun., 2017, 38, 1700582 CrossRef PubMed.
- H. M. Colquhoun and Z. Zhu, Recognition of polyimide sequence-information by a molecular tweezer, Angew. Chem., Int. Ed., 2004, 43, 5040–5045 CrossRef CAS PubMed.
- H. M. Colquhoun, Z. Zhu, C. J. Cardin, Y. Gan and M. G. B. Drew, Sterically controlled recognition of macromolecular sequence information by molecular tweezers, J. Am. Chem. Soc., 2007, 129, 16163–16174 CrossRef CAS PubMed.
- Z. Zhu, C. J. Cardin, Y. Gan and H. M. Colquhoun, Sequence-selective assembly of tweezer-molecules on linear templates enables frameshift reading of sequence information, Nat. Chem., 2010, 2, 653–660 CrossRef PubMed.
- K. Jalani, M. Kumar and S. J. George, Mixed donor–acceptor charge-transfer stacks formed via hierarchical self-assembly of a non-covalent amphiphilic foldamer, Chem. Commun., 2013, 49, 5174–5176 RSC.
- S. Ghosh and S. Ramakrishnan, Small-molecule-induced folding of a synthetic polymer, Angew. Chem., Int. Ed., 2005, 44, 5441–5447 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Supramolecular assemblies by charge-transfer interactions between donor and acceptor chromophores, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- A. J. Zych and B. L. Iverson, Synthesis and conformational characterization of tethered, self-complexing 1,5-dialkoxynaphthalene/1,4,5,8-naphthalenetetracarboxylic diimide systems, J. Am. Chem. Soc., 2000, 122, 8898–8909 CrossRef CAS.
- J. S. Shaw, R. Vaiyapuri, M. P. Parker, C. A. Murray, K. J. C. Lim, C. Pan, M. Knappert, C. J. Cardin, B. W. Greenland, R. Grau-Crespo and H. M. Colquhoun, Elements of fractal geometry in the 1H NMR spectrum of a copolymer intercalation-complex: identification of the underlying Cantor set, Chem. Sci., 2018, 9, 4052–4061 RSC.
- K.-W. Lienert, Poly(ester-imide)s for industrial use, Adv. Polym. Sci., 1999, 141, 45–82 CrossRef CAS.
- A. A. Shaikh, G. Schwarz and H. R. Kricheldorf, Macrocycles 23. Odd–even effect in the cyclization of poly(ester imide)s derived from catechols, Polymer, 2003, 44, 2221–2230 CrossRef CAS.
- T. Vlad-Bubulac, C. Hamciuc and O. Petreus, Synthesis and characterization of some polyesters and poly(ester-imide)s based on bisphenol-A derivatives, High Perform. Polym., 2010, 22, 345–358 CrossRef CAS.
- M. Hasegawa, S. Takahashi, S. Tsukuda, H. Soichi, T. Hirai, J. Ishii, Y. Yamashina and Y. Kawamura, Symmetric and asymmetric spiro-type colorless poly(ester imide)s with low coefficients of thermal expansion, high glass transition temperatures, and excellent solution-processability, Polymer, 2019, 169, 167–184 CrossRef CAS.
- M. Bruma, I. Sava, I. Negulescu, W. Daly, J. Fitch and P. Cassidy, Synthesis and properties of fluorinated poly(ester-imide)s, High Perform. Polym., 1995, 7, 411–420 CrossRef CAS.
- N. Zindy, J. T. Blaskovits, C. Beaumont, J. Michaud-Valcourt, H. Saneifar, P. A. Johnson, D. Beĺanger and M. Leclerc, Pyromellitic diimide-based copolymers and their application as stable cathode active materials in lithium and sodium-ion batteries, Chem. Mater., 2018, 30, 6821–6830 CrossRef CAS.
- C. Kulkarni and S. J. George, Carbonate linkage bearing naphthalenediimides: Self assembly and photophysical properties, Chem. – Eur. J., 2014, 20, 4537–4541 CrossRef CAS PubMed.
- B. Aradi, B. Hourahine and T. Frauenheim, DFTB+, a sparse matrix-based implementation of the DFTB method, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim and G. Seifert, Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- L. Zhechkov, T. Heine, S. Patchkovskii, G. Seifert and H. A. Duarte, An efficient a posteriori, treatment for dispersion interaction in density-functional-based tight binding, J. Chem. Theory Comput., 2005, 1, 841–847 CrossRef CAS PubMed.
- H. J. S. Smith, Proc. London Math. Soc., 1875, 6, 140–153 Search PubMed.
- M. E. Dehkordi, V. Luxami and G. D. Pantos, High-yielding synthesis of chiral donor−acceptor catenanes, J. Org. Chem., 2018, 83, 11654–11660 CrossRef CAS PubMed.
- N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantos and J. K. M. Sanders, Discovery of an organic trefoil knot, Science, 2012, 338, 783–785 CrossRef CAS PubMed.
- H. M. Colquhoun, D. J. Williams and Z. Zhu, Macrocyclic aromatic ether-imide-sulfones: versatile supramolecular receptors with extreme thermochemical and oxidative stability, J. Am. Chem. Soc., 2002, 124, 13346–13347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of polymer synthesis and characterisation; calibration plot, ηinhvs. Mn (GPC); 1H NMR titration method and data; atomic coordinates in .mol2 format. See DOI: 10.1039/c9py01460h |
| This journal is © The Royal Society of Chemistry 2019 |