
Swelling and mechanical properties of thermoresponsive/hydrophilic conetworks with crosslinked domain structures prepared from various triblock precursors†
 *a
*a
Abstract
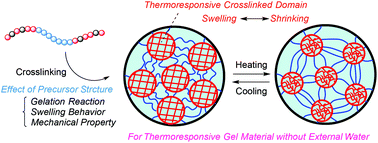
A designed amphiphilic conetwork (APCN) having thermoresponsive polymer chains is attractive for the development of novel stimuli-responsive materials with a controlled responsive behavior. We have recently proposed a novel APCN having crosslinked domain (CD) structures prepared by the post-polymerization crosslinking of controlled triblock precursor polymers with reactive sites in the outer blocks. In the current study, we evaluated the effects of the structures of the triblock precursors including the sequence, molecular weight, and composition on the gelation reaction and the swelling properties of the obtained gels in detail. The gelation reaction and the volume at the swelling state at a low temperature were strongly affected by the molecular weight of the middle block of a precursor, whereas the temperature and the sharpness of the response were controlled by the composition of a precursor. Interestingly, the gel consisting of thermoresponsive CDs and hydrophilic bridging chains had improved elastic modulus and elongation ability upon heating in air without external water, probably because water flowed between the thermoresponsive CDs and the domains of the hydrophilic bridging chains in response to temperature change.
 Please wait while we load your content...
Please wait while we load your content...

