
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of macrocyclic poly(ethylene oxide)s containing a protected thiol group: a strategy for decorating gold surfaces with ring polymers†
M. Ali
Aboudzadeh
 ab,
Mikel
Dolz
ab,
Xavier
Monnier
ab,
Estibaliz
González de San Román
c,
Daniele
Cangialosi
ab,
Marek
Grzelczak
abd and
Fabienne
Barroso-Bujans
*abde
ab,
Mikel
Dolz
ab,
Xavier
Monnier
ab,
Estibaliz
González de San Román
c,
Daniele
Cangialosi
ab,
Marek
Grzelczak
abd and
Fabienne
Barroso-Bujans
*abde
aCentro de Física de Materiales, CSIC-UPV/EHU, Paseo Manuel Lardizábal 5, 20018 Donostia-San Sebastián, Spain. E-mail: fbarroso@ehu.eus; Tel: +34 94301 8803
bDonostia International Physics Center (DIPC), Paseo Manuel Lardizábal 4, 20018 Donostia-San Sebastián, Spain
cPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda. Tolosa 72, 20018 Donostia-San Sebastián, Spain
dIKERBASQUE – Basque Foundation for Science, María Díaz de Haro 3, E-48013 Bilbao, Spain
eDepartamento de Física de Materiales, University of the Basque Country (UPV/EHU), Apartado 1072, San Sebastian 20080, Spain
First published on 19th November 2019
Abstract
We introduce an approach to synthesize macrocyclic poly(ethylene oxide)s containing a pendant protected thiol group (pSH-CPEO), which is demonstrated to be able to attach to gold surfaces without prior deprotection. Our strategy is based on a bimolecular approach by which a di-alkyne molecule derived from thiol-protected 3-mercapto-1,2-propanediol and a series of PEO bis(azides) of Mn = 2, 6, and 11 kg mol−1 are coupled via copper-catalyzed azide–alkyne cycloaddition. The cyclization reaction was verified by size exclusion chromatography and matrix-assisted laser desorption ionization time of flight mass spectrometry. In addition, we used fast scanning calorimetry to evaluate the glass transition temperature (Tg) of the synthesized pSH-CPEOs. Thanks to the extremely rapid cooling power of this technique, PEO crystallization can be circumvented and, therefore, its fully amorphous state can be investigated. The results confirmed higher Tg values for macrocycles compared to their chemically equivalent linear precursors. This result highlights the importance of the chain end in affecting the Tg of polymers. Finally, to demonstrate the ability of pSH-CPEO to covalently attach to gold surfaces, pSH-CPEO samples were allowed to react with gold-coated glass slides and the surface properties were compared with those of the samples obtained by the reaction of linear α-thiol, ω-methoxy PEO (with and without protection of the thiol group) and gold-coated glass slides. X-ray photoelectron spectroscopy data confirmed the formation of Au–S linkages as well as the removal of the thiol protector group through the quantitative analysis of the chemical composition at the surface. The contact angle data of pSH-CPEO/gold exhibited increased hydrophilicity compared to bare gold and topological effects at the interface. A reaction mechanism between the 2,4-dinitrobenzene-protected thiol group and the gold surface is also proposed.
Introduction
Macrocyclic (ring) polymers have attracted considerable interest in both fundamental polymer chemistry and polymer physics.1–3 Due to their unique topology consisting in the lack of chain ends, macrocyclic polymers differ from linear and branched polymers in terms of physical properties in solution and the molten phase. Some of these properties include the melt and intrinsic viscosity, glass transition temperature, crystallization, hydrodynamic volume, thermal and hydrolytic stability, etc.Surface modification with ring polymers is a recent attractive subject given the possibilities of the rings to lubricate the surfaces and prevent the adhesion of proteins more efficiently than linear polymers.4–10 In this field, the works developed by Benetti's group have been crucial demonstrating the superior surface properties of macrocyclic poly(2-alkyl-2-oxazoline)s (C-PAOx)s grafted onto metal oxide surfaces (TiO2 and Fe3O4),5–9 cartilage,4 and poly(glycidyl methacrylate)10 compared to linear analogs. The enhanced properties of C-PAOx-grafted surfaces, which include friction, lubrication, and antifouling when exposed to full human serum, fibrinogen, and albumin, are correlated with the absence of chain ends preventing interchain entanglements and increased grafting density compared to linear brushes.
Poly(ethylene oxide) (PEO) is the “gold standard” polymer for biomedical applications due to its biocompatibility, high water solubility, and its low immunogenicity, antigenicity, and toxicity.11 The use of PEO for modifying the surface properties of gold substrates to prevent nonspecific adsorption of proteins and to improve the colloidal stability and biocompatibility of gold nanoparticles is well recognized.12–14 Linear PEO and PEO architectures such as star-shaped, grafted, branched, hyperbranched, and dendrimer-like structures have been explored for a diversity of biomedical applications including drug delivery, wound healing, and tissue and bone engineering.11,15,16 However, only a limited amount of work has focused on the utility of macrocyclic PEO structures for biomedical applications,17–19 none of them on modifying gold substrates. In fact, in those studies, the macrocycles were not PEO homopolymers but copolymers where PEO was one of the block components imparting amphiphilicity for generating micelles.17–19 The study of macrocyclic PEO homopolymers has been mainly focused on their physical properties including their crystallization and folding20,21 and ring dynamics.22–24
Based on the inspiring results obtained by Benetti's group on C-PAOx-grafted surfaces, it is expected that cyclic PEO brushes on gold substrates also promote enhanced surface properties compared to linear PEO brushes. However, to accomplish this task, synthetic routes for making macrocyclic PEO chains containing reactive thiol groups must be developed. Macrocyclic PEO has been traditionally synthesized by the reaction of polyethylene glycol with either dichloromethane or tosyl chloride in the presence of KOH at high dilution.20,25 Allgaier et al. made an improvement of this tosyl chloride route in order to scale-up into the production of gram amounts of cyclic products.26 Other route to generate macrocyclic PEO homopolymers includes the ring closure of PEO bis(alkyne) precursors via the alkyne–alkyne Glaser intramolecular coupling.27
After click reactions emerged as an alternative to produce cyclic polymers,28 several complex architectures were able to be produced. For instance, click reactions were introduced in the fabrication of macrocyclic PEO copolymers via the copper-catalyzed azide–alkyne cycloaddition (CuAAC) intramolecular reaction of poly(ethylene glycol)-co-polycaprolactone.18 Other structures such as tadpole-shaped PEO copolymers (a cyclic PEO head connected to a linear polystyrene tail) have also been fabricated via the CuAAC bimolecular coupling between PEO bis(azide) and polystyrene bis(alkyne).29
Herein, we introduce an approach to synthesize macrocyclic PEOs containing a pendant protected thiol group (pSH-CPEO) based on a bimolecular homodifunctional approach, according to the classification given by Laurent and Grayson.30 Our method consists of the known reaction of a di-alkyne linker and PEO bis(azide) (N3-PEO-N3) via CuAAC. Our efforts were directed to produce PEO macrocycles that were reactive towards gold surfaces by incorporating a thiol group that was not reactive during the multiple synthesis steps and that easily reacted with gold surfaces. We found that by protecting the thiol group with 1-fluoro-2,4-dinitrobenzene (Sangers reagent),31 those aims were fulfilled. Moreover, we found that the obtained macrocyclic PEO samples reacted with gold substrates without the need for prior deprotection of the thiol group. These results were confirmed by surface analysis of pSH-CPEO-grafted on gold substrates (pSH-CPEO/Au) including X-ray photoelectron spectroscopy (XPS) and contact angle measurements of water droplets. Comparative samples obtained by grafting linear α-thiol, ω-methoxy PEO (with and without protection of the thiol group) on the gold substrate additionally confirmed the above results, as well as put in evidence the occurrence of topological effects on the surface properties.
Experimental part
Materials
1-Thioglycerol (≥97%,), 1-fluoro-2,4-dinitrobenzene (≥99%), triethylamine (≥99.5%), 4-pentynoic acid (95%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), PEO bis(azide) with Mn of 2 kg mol−1, 6.2 kg mol−1, and 11 kg mol−1, and copper(I) bromide (CuBr) were purchased from Sigma-Aldrich. Tetrahydrofuran, ethyl acetate, and chloroform were obtained from Scharlab, S.L. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) (≥98%) and anhydrous dichloromethane (DCM) were acquired from Acros Organics. Linear α-thiol, ω-methoxy PEO (SH-PEO-OMe) of 2, 6, and 10 kg mol−1 and α-azido, ω-methoxy PEO (N3-PEO-OMe) of 1 kg mol−1 were purchased from Sigma-Aldrich. All the chemicals were used as received without any further purification.Synthesis of 3-((2,4-dinitrophenyl)thio)propane-1,2-diol (product 2)
1-Thioglycerol (0.58 g, 5.36 mmol) and 1-fluoro-2,4-dinitrobenzene (1 g, 5.36 mmol) were dissolved in 4 mL of CHCl3 and the mixture was cooled using an ice bath. Triethylamine (1.4 mL) was slowly added to the reaction mixture under stirring. The ice bath was removed and the reaction was stirred for 17 h at room temperature. A yellow solid was precipitated during the reaction. The solid was filtered and dissolved in ethyl acetate with the aid of heating (up to 60 °C). Extraction was carried out using HCl (1 M) and then with water. The organic phase was separated, dried over anhydrous magnesium sulfate, and filtered. The solvent was removed using a rotary evaporator and the product was dried in a vacuum oven at 40 °C overnight. Finally, a residual yellow fine powder (product 2) was obtained. Yield: 70%.1H NMR (400 MHz, acetone-d6) δ 8.97 (d, J = 2.6 Hz, 1H), 8.48 (dd, J = 9.0, 2.6 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H), 4.47 (d, J = 5.3 Hz, 1H), 4.01 (dq, J = 10.1, 5.3 Hz, 2H), 3.68 (hept, J = 5.6 Hz, 2H), 3.50 (d, J = 11.5 Hz, 1H), 3.36–3.19 (m, 1H), 2.84 (s, 0H). 13C NMR (101 MHz, acetone) δ 145.2, 141.10, 129.25, 127.89, 122.02, 71.10, 66.09, 37.25. FTIR (cm−1): 3382, 3085, 2927, 1585, 1511, 1338, 1299, 1257, 1160, 1095, 1022, 917, 871, 833, 736, 671, 605.
Synthesis of 3-((2,4-dinitrophenyl)thio)propane-1,2-diyl bis(pent-4-ynoate) (product 3)
100 mg of freshly dried product 2 (0.365 mmol) were dissolved in 15 mL of anhydrous THF and cooled down to 0 °C under inert gas flow (argon). 4-Pentynoic acid (358 mg, 3.65 mmol) and EDC (623 g, 3.25 mmol) were dispersed in 15 mL of anhydrous DCM and the obtained dispersion was slowly injected into the solution of (2) under an inert atmosphere. The stirring was continued at 0 °C for 15 min, whereupon a solution of DMAP (130.38 mg, 1.07 mmol) in a small amount of DCM (<2 mL) was added to the reaction flask. The reaction proceeded for 48 h at 0 °C, and then the solvents were removed using a rotary evaporator. The obtained material was redissolved in DCM (40 mL). The DCM solution was washed with water (3 × 10 mL) and then dried over anhydrous MgSO4. MgSO4 was filtered off and the solution was concentrated under reduced pressure at 30 °C. The final product 3, an oily brown substance, was poured twice into hexane and separated by decantation. The residual oil was dried at 25 °C in a vacuum oven and stored at 5 °C before use. Yield: 70%.1H NMR (400 MHz, chloroform-d) δ 9.12 (d, J = 2.6 Hz, 1H), 8.48 (dd, J = 9.0, 2.5 Hz, 1H), 7.89 (d, J = 9.0 Hz, 1H), 5.29 (dtd, J = 7.6, 5.6, 3.4 Hz, 1H), 4.54 (dd, J = 12.2, 3.4 Hz, 1H), 4.37 (dd, J = 12.2, 5.3 Hz, 1H), 3.43 (dd, J = 14.1, 6.1 Hz, 1H), 3.30 (dd, J = 14.1, 7.5 Hz, 1H), 2.73–2.45 (m, 8H), 2.03 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 171.22, 171.10, 144.71, 144.35, 127.45, 127.14, 121.84, 82.14, 81.81, 69.50, 69.35, 69.25, 63.41, 33.18, 33.11, 32.13, 14.31. FTIR (cm−1): 3289, 3093, 2942, 1735, 1592, 1523, 1423, 1338, 1238, 1149, 1103, 1045, 917, 833, 736, 648.
Synthesis of pSH-CPEO (product 4)
0.835 mL of PMDETA (693.2 mg, 4 mmol) was slowly added to a solution of CuBr (0.574 g, 4 mmol) in anhydrous DCM (20 mL) under an inert atmosphere and the solution was stirred for 15 min. In a separate flask, a stoichiometric solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of (3) (17.37 mg, 0.04 mmol) and PEO bis(azide) 2 kg mol−1 (80 mg, 0.04 mmol) was prepared in 22 mL of anhydrous DCM. The latter solution was added to CuBr/PMDETA with the aid of a syringe pump, at a flow rate of 6 mL h−1, at 25 °C. After addition, the reaction mixture was stirred at room temperature overnight. The next day, the reaction mixture was washed with 2 M solution of ammonium chloride (3 × 60 mL) and finally with pure water. The solution was dried with MgSO4, filtered, and concentrated under reduced pressure at 30 °C using a rotary evaporator. The final product 4 was precipitated in hexane. Then, to remove chain extended products generated in intermolecular oligomerization events, the product was purified by a precipitation–fractionation procedure. First, the product was dissolved in chloroform at 50 °C. Since there were still non-soluble impurities, the solution was centrifuged and the solid impurities were removed. Heptane was then slowly added to the obtained clear chloroform solution under stirring until the solution became cloudy. Equilibrium phase separation was ensured by heating the cloudy solution until it became clear and then cooling it slowly with gentle stirring to 25 °C. The solution was centrifuged, the separated clear dilute phase was concentrated using a rotary evaporator, and the product was dried in a vacuum oven at room temperature.
:1) of (3) (17.37 mg, 0.04 mmol) and PEO bis(azide) 2 kg mol−1 (80 mg, 0.04 mmol) was prepared in 22 mL of anhydrous DCM. The latter solution was added to CuBr/PMDETA with the aid of a syringe pump, at a flow rate of 6 mL h−1, at 25 °C. After addition, the reaction mixture was stirred at room temperature overnight. The next day, the reaction mixture was washed with 2 M solution of ammonium chloride (3 × 60 mL) and finally with pure water. The solution was dried with MgSO4, filtered, and concentrated under reduced pressure at 30 °C using a rotary evaporator. The final product 4 was precipitated in hexane. Then, to remove chain extended products generated in intermolecular oligomerization events, the product was purified by a precipitation–fractionation procedure. First, the product was dissolved in chloroform at 50 °C. Since there were still non-soluble impurities, the solution was centrifuged and the solid impurities were removed. Heptane was then slowly added to the obtained clear chloroform solution under stirring until the solution became cloudy. Equilibrium phase separation was ensured by heating the cloudy solution until it became clear and then cooling it slowly with gentle stirring to 25 °C. The solution was centrifuged, the separated clear dilute phase was concentrated using a rotary evaporator, and the product was dried in a vacuum oven at room temperature.
The above procedure was repeated with higher molecular weight PEO bis(azide). In the case of LPEO6.2k, the following amounts of reagents are used: 120 mg (0.02 mmol) of PEO bis(azide), 6.2 kg mol−1; 8.69 mg (0.02 mmol) of (3); 0.287 g of CuBr (2 mmol); 0.417 mL of PMDETA (346 mg, 2 mmol), and proportional amounts of anhydrous DCM. In the case of LPEO11k, the following amounts of reagents are used: 100 mg (0.01 mmol) of PEO bis(azide), 11 kg mol−1; 4.34 mg (0.01 mmol) of (3); 0.143 g of CuBr (1 mmol); 0.210 mL of PMDETA (173.3 mg, 1 mmol), and proportional amounts of anhydrous DCM.
To provide evidence of the removal of the 2,4-dinitrobenzene group upon gold functionalization, 1 mL of product 2 in acetonitrile (c = 4 × 10−4 M) and a spoon of small gold pieces were shaken overnight in a vial. The filtered solution was monitored as a function of time by UV-Vis spectroscopy. After 24 h, the solution was dried and the product was redissolved in acetone-d6 to be analyzed by 1H NMR.
Results and discussion
Synthesis and characterization of pSH-CPEO
pSH-CPEO samples were synthesized by using a bimolecular homodifunctional approach, where a bis(alkyne) linking agent containing a protected thiol group and a series of commercially available PEO bis(azide) of Mn = 2, 6.2, and 11 kg mol−1 were coupled via CuAAC (Scheme 1). To synthesize this bis(alkyne) linker, we first protected the thiol group via the reaction of thioglycerol with 1-fluoro-2,4-dinitrobenzene.31 We then esterified the hydroxyl groups with 4-pentynoic acid in the presence of EDC/DMAP without observing thiol deprotection.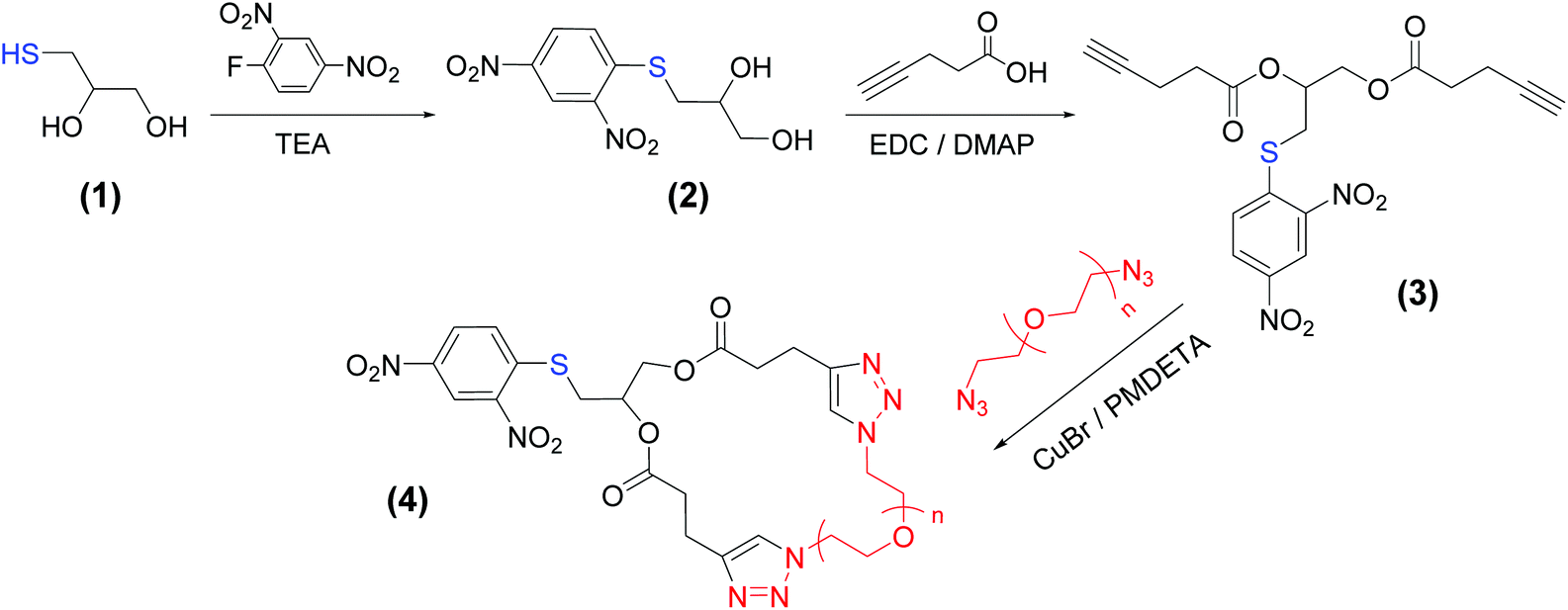 | ||
| Scheme 1 Synthesis route to pSH-CPEO. | ||
1H NMR and FTIR data (Fig. 1 and S1†) of products 2 and 3 confirm the structures obtained. 1H NMR signals corresponding to 2,4-dinitrobenzene corroborate the presence of this protector group for thiol in both products. The protection of thiol groups was also evaluated by RAMAN spectroscopy through the disappearance of S–H stretching at 2570 cm−1 in going from product 1 to 2 (Fig. 2). This signal did not appear in any product obtained in further steps of the pSH-CPEO synthesis, indicating the high stability of the 2,4-dinitrobenzene protector group during esterification (product 3) and the subsequent CuAAC click reaction (product 4).
 | ||
| Fig. 1 1H NMR data of (a) pSH-CPEO 2.4 kg mol−1 (4) (in CDCl3), (b) 3-((2,4-dinitrophenyl)thio)propane-1,2-diyl bis(pent-4-ynoate) (3) (in CDCl3), and (c) 3-((2,4-dinitrophenyl)thio)propane-1,2-diol (2) (in acetone-d6). | ||
 | ||
| Fig. 2 Raman spectra of (a) pSH-CPEO 2.4 kg mol−1 (4), (b) 3-((2,4-dinitrophenyl)thio)propane-1,2-diyl bis(pent-4-ynoate) (3), (c) 3-((2,4-dinitrophenyl)thio)propane-1,2-diol (2), and (d) 1-thioglycerol (1). | ||
Macrocyclization was then performed at high dilution by using a dropwise addition technique.28 In this case, a 1:1 equimolar solution of 3 and PEO bis(azide) was continuously dropped into a solution of Cu(I)Br and PMDETA. In this way, a very low concentration of unreacted chemical groups is ensured. To avoid intermolecular oligomerization events, the addition rate was reduced from 6 mL h−1 to 2 mL h−1 with the increasing molecular weight. After cyclization, size exclusion chromatography (SEC) data (Fig. 3) showed an increase of the retention time for all the synthesized pSH-CPEO samples in comparison to their linear precursors, as expected from the more compact structure and reduced hydrodynamic volume for macrocycles.
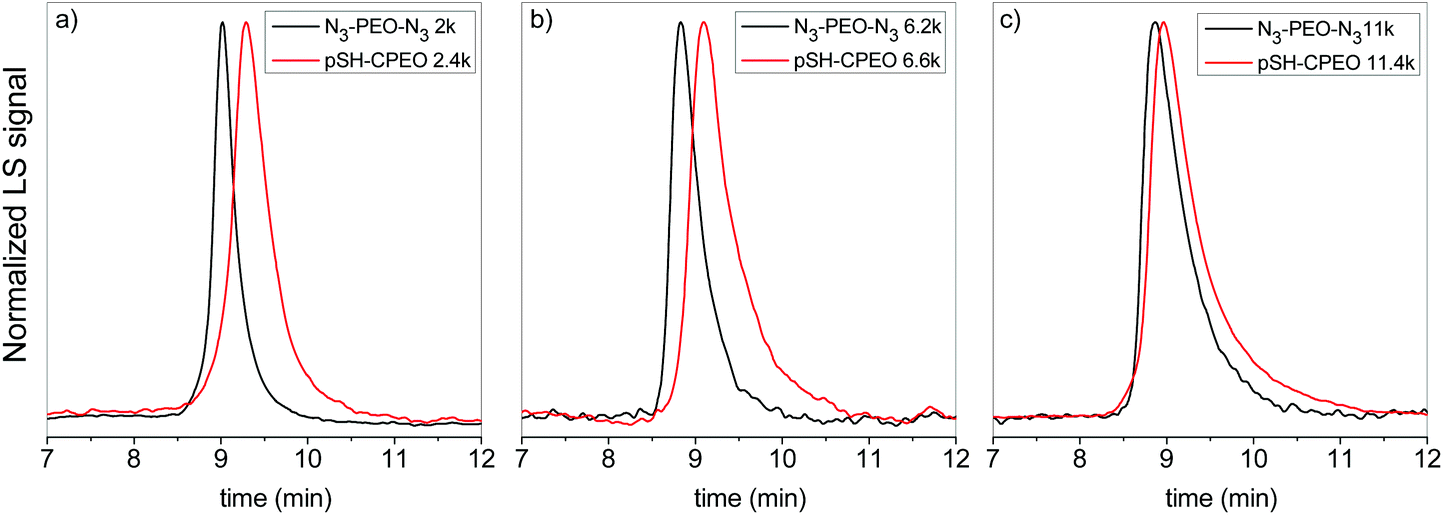 | ||
| Fig. 3 SEC data (MALS) of pSH-CPEO samples (in red) obtained from linear PEO bis(azide) (in black) of Mn = 2 kg mol−1 (a), 6.2 kg mol−1 (b), and 11 kg mol−1 (c). | ||
1H NMR data of pSH-CPEO2.4k (representative sample) exhibited the disappearance of the alkyne proton signal at 2.03 ppm in going from 3 to 4 and the appearance of a signal at about 7.64 ppm attributed to a triazole ring formed upon the CuAAC click reaction (Fig. 1). Furthermore, the FTIR data of pSH-CPEO2.4k showed the disappearance of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching band of its PEO bis(azide) precursor at 2100 cm−1, confirming the complete consumption of reactive azide groups (Fig. S1†).
N stretching band of its PEO bis(azide) precursor at 2100 cm−1, confirming the complete consumption of reactive azide groups (Fig. S1†).
MALDI-TOF MS was used to analyze the mass composition of the synthesized pSH-CPEO samples (Fig. 4). The data exhibit high intensity signals for both the macrocyclic product and the PEO bis(azide) precursor. The signals are separated by 44 Da, which corresponds to the mass of ethylene oxide repeat units. The data also revealed the formation of specimens with an overall offset of +434 Da relative to the peak distribution of starting PEO bis(azide). This mass corresponds to the mass of product 3, thus confirming the addition of a single moiety of 3 to PEO by means of the CuAAC click reaction according to Scheme 1. Similar results were observed for higher molecular weight products, indicating the viability of the synthetic method to produce pSH-CPEO samples (Fig. S2 and S3†).
 | ||
| Fig. 4 MALDI-TOF MS data of pSH-CPEO2.4k and its PEO bis(azide) precursor. The mass shift of +434 Da corresponds to the mass of (3) and that of +44 Da to the ethylene oxide monomer unit. Cation: Na+. | ||
To provide further evidence of the formation of macrocyclic PEO structures, thermal transitions of pSH-CPEO samples were evaluated by means of fast scanning calorimetry (FSC). This technique works at extremely high cooling rates (>1000 K s−1) and, therefore, allows the evaluation of glass transition temperature (Tg) of fully amorphous PEO samples, which is otherwise not possible at the standard rates of conventional DSC (<40 K min−1) due to the unavoidable crystallization of PEO. At a first glance, FSC data of Fig. 5 show notable differences in the thermal transitions of pSH-CPEO samples with respect to those of their linear precursors. The lowest molecular weight pSH-CPEO2.4k sample exhibited no crystallization at all, in contrast to its linear N3-PEO-N3 2k precursor, which exhibits cold crystallization and melting. Importantly, the pSH-CPEO2.4k sample exhibited a significant increase of the Tg of about 20 °C with respect to its linear counterpart. With the increasing molecular weight, pSH-CPEO samples crystallize after Tg (cold crystallization) but still the Tg values of the fully amorphous samples exhibit an increase with respect to their linear precursors. This Tg increment in the cyclic samples is reduced with respect to their linear precursors of higher molecular weight in 6 °C and 2 °C as depicted in Fig. 6.
 | ||
| Fig. 5 FSC curves of pSH-CPEO and linear PEO bis(azide) precursors of Mn = 2 kg mol−1 (a), 6.2 kg mol−1 (b), and 11 kg mol−1 (c). Cooling and heating rates: 1000 K s−1. | ||
 | ||
| Fig. 6 Evolution of Tg as a function of the molecular weight. Obtained at 1000 K s−1 by FSC. | ||
As known from the literature, ring polymers present higher Tg values compared to their linear analogs in the medium-low molecular weight range.2 However, the molecular weight dependence of the Tg for rings has been found to decrease, increase, or remain unaltered with the decreasing molecular weight, depending on the polymer type and the ring purity.32–34 For instance, macrocyclic poly(dimethylsiloxane) showed increasing Tg values with the decreasing molecular weight,35,36 in agreement with the theoretical model of Di Marzio and Guttman.37 In contrast, macrocycles of polystyrene,38 poly(2-vinylpyridine),38 poly(α-methylstyrene),38 poly (2-vinylnaphthalene),38 poly(phenylmethylsiloxanes),39 and poly(glycidyl phenyl ether)40 exhibited decreasing Tg values with the decreasing molecular weight. This is also the case for cyclic PEO according to the data recovered from the literature on amorphized samples of low molecular weight41,42 and plotted in Fig. S4.†
It is worthy to note that in our macrocyclic polymer samples, the mass contribution of moiety 3 into pSH-CPEO is much higher as the molecular weight of the PEO fragment is lower. These values present a variation of 20, 7, and 4 wt% for PEO Mn of 2, 6.2, and 11 kg mol−1, respectively. The presence of an organic moiety with a mass of 434 g mol−1 and a different chemical structure from that in PEO is likely to be the responsible for the Tg increase with decreasing Mn. This hypothesis is supported by the opposite trend observed in the Tg values of cyclic PEO samples composed only of ethylene oxide units (Fig. S4†). Moreover, a two-arm linear PEO (pSH-2armPEO) with a similar composition to pSH-CPEO 2.4k was synthesized by the reaction of product 3 and two equivalents of N3-PEO-OMe of 1 kg mol−1 (Scheme S1†). The higher Tg value obtained for this product compared to N3-PEO-N3 supports that moiety 3 contributes to the slower segmental dynamics observed for both pSH-2armPEO and pSH-CPEO samples. However, the much higher Tg value still exhibited by pSH-CPEO compared to pSH-2armPEO indicates that the cyclic topology, lacking of relatively fast chain ends, is the predominant factor in the distinct Tg behavior. Hence, our results allow quantifying the effect of chain ends on the decrease of Tg. In this case, the observation of Fig. 6 provides a Tg difference between cyclic and linear PEOs of the analogous chemical structure and a molecular weight of about 13 K. This difference is analogous to that found on other flexible polymer, PDMS, at a similar molecular weight.36
The crystallization and melting behavior of pSH-CPEO samples were also different from those of their linear precursors. Crystallization was avoided in pSH-CPEO2.4k unlike in its equivalent pSH-2armPEO sample, which exhibits a cold crystallization peak at 10.6 °C (Fig. S5†). We attribute this behavior to the cyclic topology, which hinders chain folding. In pSH-CPEO samples of 6.6 and 11.4 kg mol−1, we found higher cold crystallization temperature (Tc) values but lower melting temperatures (Tm) than in their linear precursors (Fig. 5 and S6†). These differences can be probably attributed also to the cyclic topology, since in these two samples, the mass contribution of 3 in the polymer is of only 7 and 4 wt%. The lower Tm values (out of equilibrium) found for the rings are in agreement with previous work of Floudas et al.21 on the study of the crystallization behavior of macrocyclic PEO by using standard DSC. It is important to note that our melting/crystallization temperatures are not comparable with their values since the cooling/heating rates used in both studies are different by a factor of 6000.
Decoration of gold surfaces with pSH-CPEO
Finally, to demonstrate the ability of pSH-CPEO to covalently attach to gold surfaces without needing the prior deprotection of the thiol group, pSH-CPEO samples were allowed to react with gold-covered glass slides by soaking the gold substrate into an aqueous solution of pSH-CPEO (Scheme 2). For comparison, similar experiments were performed with α-thiol, ω-methoxy PEO with and without protection of the thiol group (SH-PEO-OMe and pSH-PEO-OMe, respectively). The obtained samples were characterized by XPS and contact angle measurements with water droplets. | ||
| Scheme 2 Surface functionalization of gold substrates with (a) pSH-CPEO and (b) SH-PEO-OMe and pSH-PEO-OMe. | ||
Fig. 7 shows the S 2p core level spectra of pSH-CPEO6.6k and modified gold substrates with pSH-CPEO and SH-PEO-OMe samples. The S 2p core level appears as a doublet because of the spin orbit coupling. The doublet with the 2p3/2 component at binding energies of 162.1 eV for SH-PEO-OMe/Au and 162.5 eV for pSH-CPEO/Au is attributed to Au–S linkage, whereas the doublet with the 2p3/2 component located at a binding energy of about 164 eV is attributed to S not bonded to Au (in this case of S–C linkage).43,44 The spectrum of the not bonded cyclic polymer is dominated by the S–C component, as expected. The peak position for the S 2p3/2 components for all the samples is reported in Table S1†, and the quantitative analysis of the chemical compositions is reported in Table 1. The data show that the not bonded cyclic polymer (pSH-CPEO6.6k) contains a nitrogen to sulfur atomic ratio (N/S) of 15.5. This amount is reduced to 3.8 in the pSH-CPEO6.6k/Au sample confirming that the polymer has been covalently linked to the gold substrate via the removal of 2,4-dinitrobenzene moieties and the formation of S–Au linkages. The reduction of the N/S value obtained for pSH-PEO-OMe after the reaction with the gold substrate also supports the above conclusion.
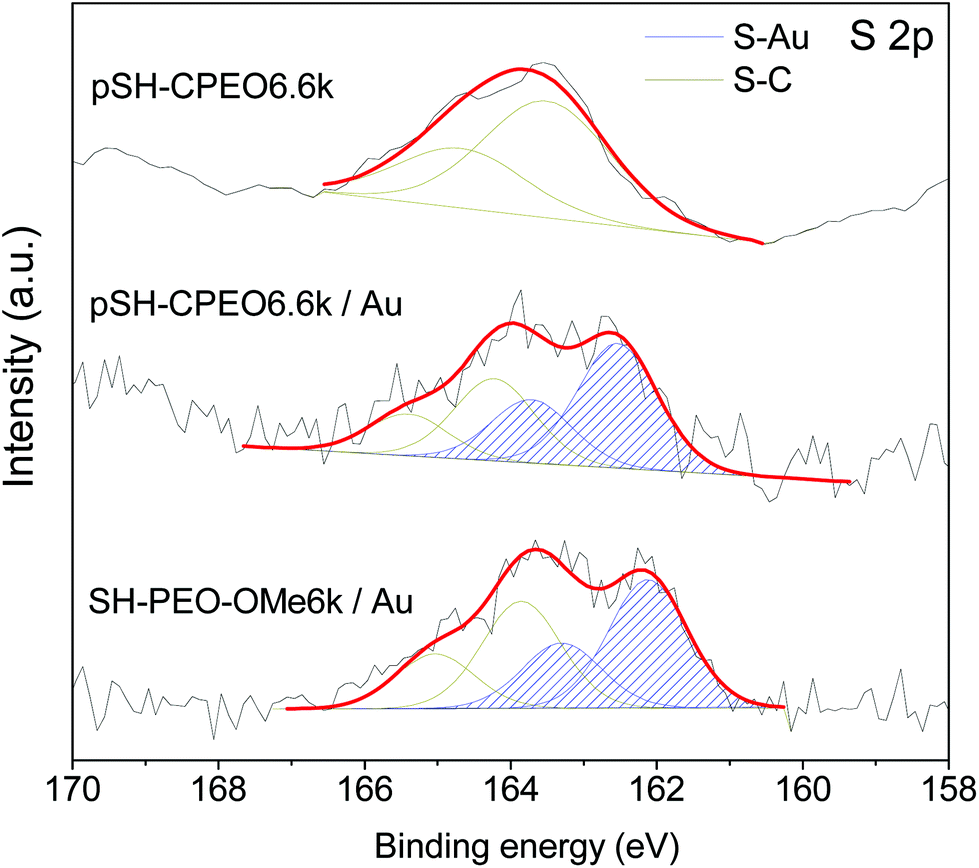 | ||
| Fig. 7 XPS data of the S 2p core level and corresponding fitted curves (S 2p3/2 and S 2p1/2 peaks are separated by 1.2 eV). The spectral intensity of the S 2p doublet for S–Au linkage is blue shaded. | ||
| Sample | Au (at%) | C (at%) | N (at%) | S (at%) | O (at%) | N/S | Sulfur coveragea (atoms per cm2) |
|---|---|---|---|---|---|---|---|
| a Calculated according to ref. 45 and 46. | |||||||
| pSH-CPEO6.6k | — | 82.6 | 3.1 | 0.2 | 14.2 | 15.5 | — |
| pSH-CPEO6.6k/Au | 31.3 | 43.2 | 1.9 | 0.5 | 17.9 | 3.8 | 6.8 × 1013 |
| pSH-PEO-OMe6.2k | — | 98.0 | 1.4 | 0.6 | 24.6 | 2.3 | — |
| pSH-PEO-OMe6.2k/Au | 42.2 | 54.2 | 1.7 | 1.9 | 14.1 | 0.9 | 4.8 × 1013 |
| SH-PEO-OMe6k/Au | 31.6 | 47 | — | 0.6 | 20.8 | — | 8.1 × 1013 |
An estimation of the grafting density can be performed by analysis of the surface coverage obtained from the sulfur to gold atomic ratios.45 The results exhibit a higher value for the cyclic structure than for its equivalent linear compound, pSH-PEO-OMe/Au, both containing the protected thiol group. These results are in agreement with the results obtained by Benetti et al., who observed higher grafting densities for cyclic brushes than for linear ones.5,6
The effect of the 2,4-dinitrobenzene protecting group on the efficiency of gold functionalization was evaluated by comparing the surface coverage values in both linear samples with protected and unprotected thiol groups (pSH-PEO-OMe/Au vs. SH-PEO-OMe/Au, respectively). The results showed a lower value for the sample containing a protected thiol group, which we attributed to the bulkier 2,4-dinitrobenzene group demanding more space to reach the surface, or that the reaction kinetics is slightly slower compared to that of unprotected thiol. Nonetheless, the advantages of using a protected thiol group for surface modification (easier manipulation and storage) outweigh this disadvantage.
The contact angle (CA) measurements of polymer-functionalized gold surfaces and bare gold substrate are reported in Table 2. The CA obtained for the unmodified gold is similar to that reported by other groups.47 As observed, the gold substrate became much more hydrophilic when modified with pSH-CPEO indicating successful functionalization of the metal surface. Similarly, the modification of the gold substrate with SH-PEO-OMe and pSH-PEO-OMe led to reduced CA values compared to that of the gold substrate, as expected from the high hydrophilicity of PEO chains. Moreover, linear chains with protected and unprotected thiol groups exhibited similar CA values and a slight increase with the molecular weight. Based on the distinct surface coverage previously observed for both samples, we can conclude that the CA of water droplets on these types of samples is not much sensitive to the grafting density. Interestingly, gold surfaces modified with pSH-CPEO exhibited increased CA values with respect to linear analogs and increased differences between cyclic and linear analogs with the increasing molecular weight. These differences can be originated from both the distinct topologies and due to the presence of moiety 3 in the cyclic samples. The latter effect was evaluated by measuring the CA value of a gold surface modified with pSH-2armPEO (Mn = 2.4 g mol−1), which resulted in a value of 33 ± 2°, similar to that obtained in the analogous cyclic sample. It is likely that the presence of moiety 3 promotes increasing hydrophobicity; however, this effect should decrease with the increasing molecular weight. Therefore, we attribute the higher CA values for the gold surfaces modified with pSH-CPEO compared to linear analogs to a topological effect on the surface wettability. Similar results were previously observed by Grubbs et al. in thin films of cyclic and linear polyethylenes.48 They observed a higher CA value for the cyclic polymer, with a difference of 9 ± 2° with respect to that of the linear analog. They explained these results as an effect of the migration of the linear polymer's end group to the surface. This effect is absent in the cyclic polymer as well as in higher molecular weight samples (as they also observed), thus confirming the existence of topological effects at the interface.
| M n (kg mol−1) | CA (pSH-CPEO) (°) | CA (pSH-PEO-OMe) (°) | CA (SH-PEO-OMe) (°) |
|---|---|---|---|
| 2 | 32 ± 1 | 25 ± 1 | 28 ± 2 |
| 6 | 41 ± 2 | 32 ± 1 | 31 ± 2 |
| 11 | 45 ± 1 | 33 ± 1 | 34 ± 1 |
To provide further evidence of the removal of the 2,4-dinitrobenzene group upon gold functionalization, a diluted solution of product 2 in acetonitrile (no water soluble) and small gold pieces were allowed to react for 24 h. The reaction was monitored by UV-Vis spectroscopy, which exhibited the appearance of a shoulder at about 300 nm after 24 h of reaction (Fig. S7†). Then, the solution was filtered off, and the product was dried and analyzed by 1H NMR and 1H–1H COSY NMR (Fig. 8 and Fig. S8,† respectively). The aromatic region of the spectrum showed the presence of unreacted product 2, as expected from its excess amount with respect to the gold surface area, and the appearance of peaks that indicate the formation of reaction products. Total structural identification of the formed molecules has not been possible, although their peak position and COSY data suggest that they are composed of 2,4-dinitrobenzene moieties and that their structure is different from that of starting product 2. The results suggest that gold reacts with the protected thioether, likely assisted by a nucleophile, leading to the formation of a Au–S linkage and the release of 2,4-dinitrobenzene moiety (Scheme 3).
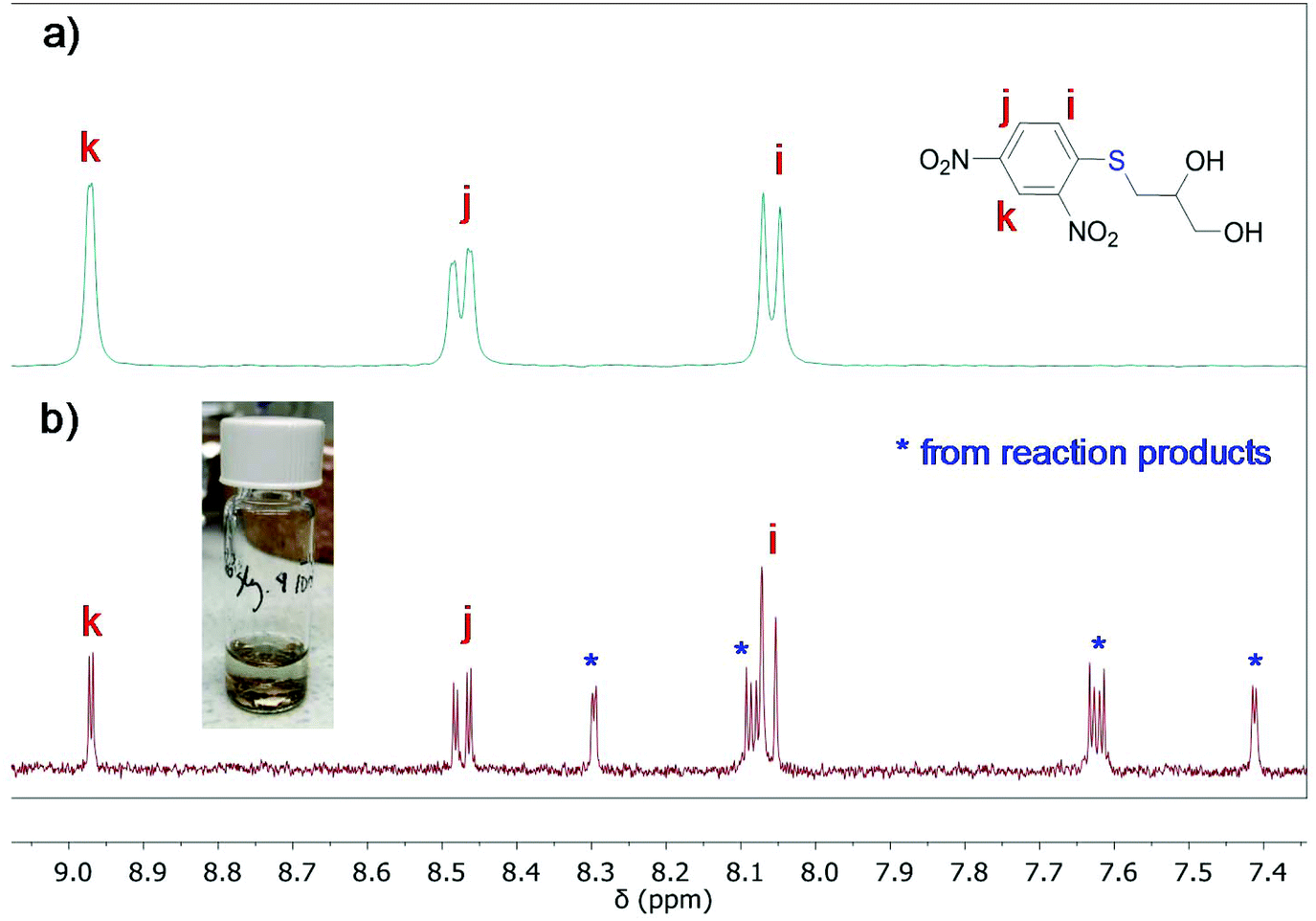 | ||
| Fig. 8 1H NMR (acetone-d6) of (a) starting product 2 and (b) product 2 after the reaction with gold metal pieces in acetonitrile for 24 h. | ||
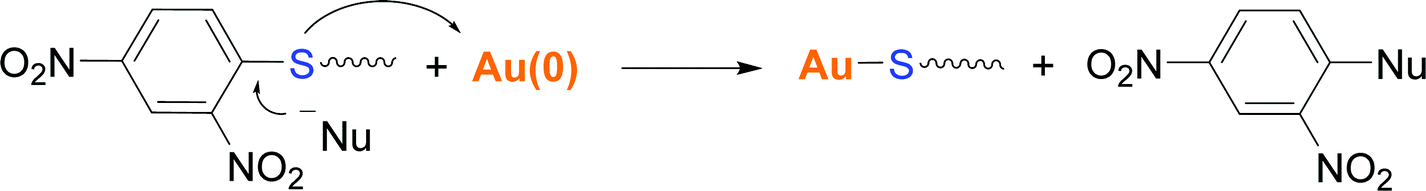 | ||
| Scheme 3 The proposed reaction mechanism of gold modification with the 2,4-dinitrobenzene-protected thiol group. | ||
Conclusions
Macrocyclic poly(ethylene oxide)s containing a pendant protected thiol group have been synthesized by the click reaction of PEO bis(azide) with a bis(alkyne) linker previously designed and synthesized by us. This linker provides the cyclic polymer with a thiol group protected with 2,4-dinitrobenzene, which is stable under conditions of esterification and the CuAAC reaction. Moreover, the thiolated macrocyclic poly(ethylene oxide)s are able to covalently attach to a gold surface without the need for prior deprotection of the thiol group. This method is advantageous since it allows an easy manipulation and storage of thiolated compounds before usage.Owing to the proved differences in the physical and chemical properties between the ring and linear polymers observed in the past and in current study in terms of the hydrophilicity of modified gold surfaces, we expect that our approach can also be used to address fundamental questions related to polymer topology at the surface. From a broader perspective, we foresee the use of this system as a means of decorating gold nanoparticles with potential applications in biomedical devices.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Farihah M. Haque for helpful discussions on MALDI-TOF MS and Dr. J. I. Miranda for experiments and helpful discussions on NMR. We also gratefully acknowledge support from the Spanish Ministry “Ministerio de Ciencia, Innovación y Universidades” (PGC2018-094548-B-I00, MICINN/FEDER, UE), Basque Government (IT-1175-19 and PIBA 2018-34), and Diputación Foral de Guipúzcoa (RED 2018).References
- J. A. Semlyen, Cyclic Polymers, Kluwer Academic Publishers, New York, Boston, Dordrecht, London, Moscow, 2nd edn, 2002 Search PubMed.
- Y. Tezuka, Topological Polymer Chemistry. Progress of Cyclic Polymers in Syntheses, Properties and Functions, Word Scientific Publishing Co. Pte. Ltd., 2013 Search PubMed.
- D. Richter, S. Gooßen and A. Wischnewski, Soft Matter, 2015, 11, 8535–8549 RSC.
- G. Morgese, E. Cavalli, J.-G. Rosenboom, M. Zenobi-Wong and E. M. Benetti, Angew. Chem., Int. Ed., 2018, 57, 1621–1626 CrossRef CAS PubMed.
- G. Morgese, B. Shirmardi Shaghasemi, V. Causin, M. Zenobi-Wong, S. N. Ramakrishna, E. Reimhult and E. M. Benetti, Angew. Chem., Int. Ed., 2017, 56, 4507–4511 CrossRef CAS PubMed.
- G. Morgese, L. Trachsel, M. Romio, M. Divandari, S. N. Ramakrishna and E. M. Benetti, Angew. Chem., Int. Ed., 2016, 55, 15583–15588 CrossRef CAS PubMed.
- M. Divandari, G. Morgese, S. N. Ramakrishna and E. M. Benetti, Eur. Polym. J., 2019, 110, 301–306 CrossRef CAS.
- M. Divandari, G. Morgese, L. Trachsel, M. Romio, E. S. Dehghani, J.-G. Rosenboom, C. Paradisi, M. Zenobi-Wong, S. N. Ramakrishna and E. M. Benetti, Macromolecules, 2017, 50, 7760–7769 CrossRef CAS.
- M. Divandari, L. Trachsel, W. Yan, J.-G. Rosenboom, N. D. Spencer, M. Zenobi-Wong, G. Morgese, S. N. Ramakrishna and E. M. Benetti, ACS Macro Lett., 2018, 7, 1455–1460 CrossRef CAS.
- W. Yan, M. Divandari, J.-G. Rosenboom, S. N. Ramakrishna, L. Trachsel, N. D. Spencer, G. Morgese and E. M. Benetti, Polym. Chem., 2018, 9, 2580–2589 RSC.
- C. Dingels and H. Frey, in Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II, ed. V. Percec, Springer International Publishing, Cham, 2013, pp. 167–190, DOI:10.1007/12_2013_235.
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- E. Polo, V. Araban, B. Pelaz, A. Alvarez, P. Taboada, M. Mahmoudi and P. del Pino, Appl. Mater. Today, 2019, 15, 599–604 CrossRef.
- M. G. Soliman, B. Pelaz, W. J. Parak and P. del Pino, Chem. Mater., 2015, 27, 990–997 CrossRef CAS.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- S. Honda, T. Yamamoto and Y. Tezuka, Nat. Commun., 2013, 4, 1574 CrossRef PubMed.
- B. Zhang, H. Zhang, Y. Li, J. N. Hoskins and S. M. Grayson, ACS Macro Lett., 2013, 2, 845–848 CrossRef CAS.
- E. Baba, T. Yatsunami, Y. Tezuka and T. Yamamoto, Langmuir, 2016, 32, 10344–10349 CrossRef CAS PubMed.
- J. Cooke, K. Viras, G.-E. Yu, T. Sun, T. Yonemitsu, A. J. Ryan, C. Price and C. Booth, Macromolecules, 1998, 31, 3030–3039 CrossRef CAS.
- G. Zardalidis, J. Mars, J. Allgaier, M. Mezger, D. Richter and G. Floudas, Soft Matter, 2016, 12, 8124–8134 RSC.
- S. Nam, J. Leisen, V. Breedveld and H. W. Beckham, Polymer, 2008, 49, 5467–5473 CrossRef CAS.
- S. Gooßen, A. R. Brás, M. Krutyeva, M. Sharp, P. Falus, A. Feoktystov, U. Gasser, W. Pyckhout-Hintzen, A. Wischnewski and D. Richter, Phys. Rev. Lett., 2014, 113, 168302 CrossRef PubMed.
- A. R. Brás, S. Gooßen, M. Krutyeva, A. Radulescu, B. Farago, J. Allgaier, W. Pyckhout-Hintzen, A. Wischnewski and D. Richter, Soft Matter, 2014, 10, 3649–3655 RSC.
- G.-E. Yu, P. Sinnathamby, C. Price and C. Booth, Chem. Commun., 1996, 31–32 RSC.
- C. H. Hövelmann, S. Gooßen and J. Allgaier, Macromolecules, 2017, 50, 4169–4179 CrossRef.
- Y. Zhang, G. Wang and J. Huang, Macromolecules, 2010, 43, 10343–10347 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS PubMed.
- Y. Tao and H. Zhao, Polymer, 2017, 122, 52–59 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- G. Carrot, J. G. Hilborn, M. Trollsås and J. L. Hedrick, Macromolecules, 1999, 32, 5264–5269 CrossRef CAS.
- L. Gao, J. Oh, Y. Tu, T. Chang and C. Y. Li, Polymer, 2019, 170, 198–203 CrossRef CAS.
- Y. Gan, D. Dong and T. E. Hogen-Esch, Macromolecules, 1995, 28, 383–385 CrossRef CAS.
- P. G. Santangelo, C. M. Roland, T. Chang, D. Cho and J. Roovers, Macromolecules, 2001, 34, 9002–9005 CrossRef CAS.
- S. J. Clarson, K. Dodgson and J. A. Semlyen, Polymer, 1985, 26, 930–934 CrossRef CAS.
- K. U. Kirst, F. Kremer, T. Pakula and J. Hollingshurst, Colloid Polym. Sci., 1994, 272, 1420–1429 CrossRef CAS.
- E. A. Di Marzio and C. M. Guttman, Macromolecules, 1987, 20, 1403–1407 CrossRef CAS.
- T. E. Hogen-Esch, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2139–2155 CrossRef CAS.
- S. J. Clarson, J. A. Semlyen and K. Dodgson, Polymer, 1991, 32, 2823–2827 CrossRef CAS.
- T. Gambino, A. Martínez de Ilarduya, A. Alegría and F. Barroso-Bujans, Macromolecules, 2016, 49, 1060–1069 CrossRef CAS.
- S. Singla and H. W. Beckham, Macromolecules, 2008, 41, 9784–9792 CrossRef CAS.
- F. Barroso-Bujans and A. Alegria, Phys. Chem. Chem. Phys., 2017, 19, 18366–18371 RSC.
- J. F. Moulder, J. Chastain and R. C. King, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics, 1995 Search PubMed.
- NIST, X-ray Photoelectron Spectroscopy Database, NIST Standard Reference Database Number 20, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2000, DOI:10.18434/T4T88K, (retrieved 27 June 2019).
- E. Oh, K. Susumu, A. J. Mäkinen, J. R. Deschamps, A. L. Huston and I. L. Medintz, J. Phys. Chem. C, 2013, 117, 18947–18956 CrossRef CAS.
- M. P. Seah, Surf. Interface Anal., 2012, 44, 1353–1359 CrossRef CAS.
- J. Canning, N. Tzoumis, J. K. Beattie, B. C. Gibson and E. Ilagan, Chem. Commun., 2014, 50, 9172–9175 RSC.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization techniques and supplementary data of NMR, FTIR, Raman spectroscopy, MALDI-TOF MS, FSC and XPS. See DOI: 10.1039/c9py01394f |
| This journal is © The Royal Society of Chemistry 2019 |