
Recyclable sulfone-containing polymers via ring-opening polymerization of macroheterocyclic siloxane monomers: synthesis, properties and recyclability†
 *a
and
Shengyu
Feng
*a
*a
and
Shengyu
Feng
*a
Abstract
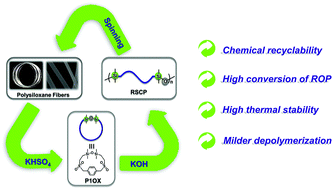
Environmental pollution and the dependence on finite resources are intensifying the search for new chemically recyclable polymers. Herein, we report the synthesis and properties of recyclable sulfone-containing polysiloxanes via the anionic ring-opening polymerization (ROP) of a new macroheterocyclic siloxane monomer (P1OX). The resulted polymer with a molecular weight up to 49 kg mol−1 shows good thermal stability and chemical recyclability. Compared to previous recyclable polymers, the recyclable polysiloxanes have simpler synthetic methods without expensive catalysts, milder conditions of depolymerization (using available and cheap KHSO4 at ambient temperature) and high selectivity of depolymerization (92%) back to P1OX. The sulfone-containing polysiloxanes were a brownish rigid plastic with a storage modulus E′ of 203 MPa at room temperature. From these recycled polymers, smooth polysiloxane nanofibers were prepared successfully via electrospinning. These results conclusively indicated that the new family of sulfone-containing polysiloxanes largely extended the scope of application of silicone and sustainable materials.
 Please wait while we load your content...
Please wait while we load your content...

