
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereocomplexation of cyclic polylactides with each other and with linear poly(L-lactide)s†
Andreas
Meyer
a,
Steffen M.
Weidner
 b and
Hans R.
Kricheldorf
*c
b and
Hans R.
Kricheldorf
*c
aInstitute of Physical Chemistry, University of Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany
bFederal Institute for Materials Research and Testing – BAM, Richard Willstätter Str. 11, D-12489 Berlin, Germany
cInstitute of Technical und Macromolecular Chemistry, Bundesstr. 45, D-20146 Hamburg, Germany. E-mail: hrkricheldorf@aol.de
First published on 28th October 2019
Abstract
Two kinds of cyclic poly(D- and L-lactide)s were synthesized, namely CI labeled samples mainly consisting of even-numbered cycles with low dispersity and CII, CIII or CIV-labeled ones consisting of equal amounts of even and odd-numbered cycles with high dispersity and higher molecular weights (Mw up to 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Furthermore, linear poly(L-lactide)s were prepared by initiation with ethanol and in both series the molecular weight was varied. The formation of stereocomplexes from cyclic poly(D-lactide)s and all kinds of poly(L-lactide)s was performed in dichloromethane/toluene mixtures. The stereocomplexes crystallized from the reaction mixture were characterized in the virgin state and after annealing at 205 °C. Stereocomplexes free of stereohomopolymers with crystallinities up to 80% were obtained from all experiments in yields ranging from 60 to 80%. Despite the high annealing temperature (maintained for 1 h), little transesterification was observed and the crystallinity slightly increased.
000). Furthermore, linear poly(L-lactide)s were prepared by initiation with ethanol and in both series the molecular weight was varied. The formation of stereocomplexes from cyclic poly(D-lactide)s and all kinds of poly(L-lactide)s was performed in dichloromethane/toluene mixtures. The stereocomplexes crystallized from the reaction mixture were characterized in the virgin state and after annealing at 205 °C. Stereocomplexes free of stereohomopolymers with crystallinities up to 80% were obtained from all experiments in yields ranging from 60 to 80%. Despite the high annealing temperature (maintained for 1 h), little transesterification was observed and the crystallinity slightly increased.
Introduction
The formation of a so-called stereocomplex of poly(D-lactide) and poly(L-lactide) was first reported by Ikada and co-workers in 1987.1 This research group has continued to explore syntheses and properties of polylactide stereocomplexes (StCs) during the following years2–9 and one member of this group, H. Tsuji, has continued to work on the characterization of StCs until recent years.10–14 Characteristic for these StCs is a different crystal lattice (a triclinic cell called modification) and a melting point in the range of 228–231 °C which is 50 °C higher than that of an annealed optically pure poly(D-lactide) or poly(L-lactide). The term stereocomplex is, in principle, misleading, because its architecture is nothing else than a racemate of two enantiomeric isotactic polymers quite analogous to the racemate of the cyclic monomer i.e.D,D,L,L-lactide. The intermolecular forces that hold the enantiomeric chains together are van der Waals forces and dipole–dipole interactions, which in the case of the CH3–O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interaction may approach the quality of a weak H-bond.15 Yet, the ligand–metal atom bonds typical for a true complex are not involved. However, the term stereocomplex (StC) is widely used in the literature and thus, also used in this work.
C interaction may approach the quality of a weak H-bond.15 Yet, the ligand–metal atom bonds typical for a true complex are not involved. However, the term stereocomplex (StC) is widely used in the literature and thus, also used in this work.
Since 1993 the work of Ikada and co-workers has been expanded by other research groups and properties of such StCs were explored in various directions, summarized and discussed by various authors in several review articles.16–20 In addition to StCs based on two separate enantiomeric chains,1–9,15,21–24 various types of block-copolymers were prepared and characterized. These block copolymers may be subdivided into two categories. The first category are the stereo-blockcopolymers which contain blocks of D- and L-units in the same chain. D/L-ratio and block lengths were varied over a broad range. Polylactides having a random or nearly random sequence of D- and L-units cannot crystallize and do not form stereocomplexes. However, above a certain block length StC formation can occur and both, Tm and melting enthalpy (ΔHm) increase with length and perfection of the D- or L-blocks.25–38 The second category consists of two- or three-block copolymers containing blocks of different chemical structure, such as blocks of poly(e-caprolactone), poly(ethylene glycol) or poly(dimethyl siloxane).
More recently, two research groups reported on attempts to prepare and characterize StCs of cyclic polylactides. Tezuka and coworkers synthesized the cycles by a ring-closing reaction of linear, two-block copolylactides consisting of one L-block and one D-block in combination with functional end groups.39,40 Yet, in this way guest–host copolymers and not cyclic homopolymers were obtained. Depending on whether the two blocks were arranged in a parallel or antiparallel fashion Tm values of 221 °C or 203 °C were found.
Waymouth and coworkers prepared cyclic and linear poly(L-lactide)s and poly(D-lactide)s by means of a heterocyclic carbine catalyst, which due to its basicity caused partial racemization.41 The optical rotation data suggest that the cycles contained approximately 40% of D-units and the linear chains approximately 25%. For the combination of enantiomeric cycles a Tm of 179 °C and ΔHm 15 J g−1 was determined, whereas a combination of cyclic with linear enantiomers resulted in Tm values of 178 or 185 °C with ΔHm of 20 or 41 J g−1.
In this context it was the purpose of the present work to elucidate to what extent optically pure cyclic polylactides prepared by various tin catalysts (Scheme 1) can form stereocomplexes with each other or with linear polylactides of similar molecular weight.42–47 Four different cyclic polylactides were used for this study that varied both by their weight average molecular weight (Mw) in the range of 10000 to 300000 g mol−1, and by their dispersities.
 | ||
| Scheme 1 Catalysts used for the preparation of the cyclic poly(D-lactide)s and poly(L-lactide)s. | ||
Experimental
Materials
D- and L-lactide (originating from Carbion PURAC) were kindly supplied by Thyssen Uhde AG (Berlin) and recrystallized from “toluene 99.89% extra dry” of ACROS Organics (Geel, Belgium). The syntheses of the catalysts BuSnOPF, SnNaph2 and SnBiph were described previously.42–45 Dichloromethane was distilled over P4O10 and chlorobenzene was distilled over P4O10in vacuo.Polymerizations
CI-L was prepared analogously, but monomer and catalyst were dissolved in 42 mL of chlorobenzene (1 M solution).
Syntheses of stereocomplexes
D-Polylactide (2 g) and a L-polylactide (2 g) were dissolved in 50 mL of dichloromethane at 20–22 °C. Afterwards the resulting solution was thermostated at 50 °C for 30 min. Toluene (25 mL) was then added (the reaction mixture still remained clear). The temperature was raised to 70 °C and maintained for approx. 30 min, whereupon part of the dichloromethane distilled off. The temperature was raised to 90 °C for additional 30 min, and more dichloromethane was removed. Toluene (10 mL) was added and the temperature was raised to 15 °C, whereupon part of the stereocomplex began to precipitate. Finally, the temperature was raised to 135 °C and part of the toluene was distilled off. The remaining suspension of stereocomplex in approx. 25 mL of toluene was diluted with dichloromethane (50 mL) and stirred for 20 min to dissolve unreacted enantiomers if present. The product was then isolated by filtration, washed with dichloromethane and dried at 58 °C in vacuo for 24 h.Measurements
The 600 MHz 1H NMR spectra were recorded with a Bruker Avance 600 FT NMR spectrometer in 5 mm sample tubes. CDCl3 containing TMS served as solvent and for shift referencing. The DSC measurements were performed on a Mettler Toledo DSC-1, equipped with the Stare Software version 10.1, at a heating rate of 10 K min−1. Only the first heating trace was evaluated. The WAXS measurements were performed using our in-house SAXS/WAXS apparatus equipped with an Incoatec™ X ray source IμS and Quazar Montel optics. The wavelength of the X ray beam was 0.154 nm and the focal spot size at the sample position was 0.6 mm2. The samples were measured in transmission geometry. The sample-detector distance was 0.1 m, allowing us to detect an angular range of 2Θ = 5°–33°. The patterns were recorded with a Rayonix™ SX165 CCD-Detector and the accumulation time per WAXS measurement was 600 s. DPDAK; a customizable software for reduction and analysis of X-ray scattering data sets was used for gathering 1D scattering curves.46 For the evaluation of the crystallinity of the samples the data were imported in Origin™ and analyzed with the curve fitting module. After subtracting of the instrumental background, the integral intensity of the crystalline reflections was divided by the overall integral intensity to determine the crystallinity xc.MALDI TOF mass spectra were recorded on an Autoflex III Smartbeam (Bruker Daltonik, Bremen, Germany). Spectra were recorded in linear mode at 200 Hz. The matrix solution (DCTB – trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile, 10 mg mL−1) was doped with 5 μL of potassium trifluoroacetate (KTFA 2 mg ml−1) to selectively enable the formation of potassium adduct ions. 20 μL of dissolved samples (5 mg mL−1, chloroform) were mixed with 50 μL of the matrix solution. Finally, 1 μL of this solution was deposited on a stainless-steel target. The manufactures software (FlexControl and FlexAnalysis) was applied for recording and evaluation. Typically, 2000 single spectra recorded on different positions were accumulated for one spectrum.
SEC measurements were performed using a modular system consisting of an isocratic pump (Chromophor C 1130) running at 1 mL min−1 flow rate. The system was kept at 40 °C. Samples with a concentration of 2–4 mg mL−1 were manually injected by a Rheodyne valve. The sample volume was 100 μL. A triple detector combination was used consisting of a refractive index detector RI-501 (Shodex), a multi angle laser light scattering detector DAWN and a viscometer Viscostar (both Wyatt Germany, Dernbach). Clarity software (GPC extension, DataApex) was used for instrument control and molecular weight calculation, whereas ASTRA 6 (Wyatt) was used to perform the MHS calculations. The SEC instrument calibration was performed using commercially available polystyrene standard sets (Polymer Standards Service – PSS, Mainz, Germany). Due to peaks caused by solvent and low mass impurities, the calculation of the number average (Mn) and weight average (Mw) molecular weights and the dispersities Đ started from 1100 g mol−1 onwards. Therefore, probably not all oligomers were included in the calculation. This difference has only little influence on Mw, whereas Mn values may be too low, and thus, dispersities might be correspondingly underestimated.
Results and discussion
Preparation of starting materials
Four different kinds of cyclic polylactides were prepared and labelled CI-CIV, whereupon the roman number increases with the molecular weight (see Table 1).| Label | Even/odd | M n | M w | Đ | |
|---|---|---|---|---|---|
| a Measured after precipitation into ligroin. | |||||
| CI-D | 9/1 |
|
12700 |
1.1 | |
| CIII-D | 1/1 | 26500 |
82500 |
3.1 | |
| CI-L | 9/1 | 1100 | 12200 |
1.1 | |
| CII-L | 1/1 | 10500 |
27500 |
2.6 | |
| CIII-L | 1/1 | 19500 |
63000 |
3.1 | |
| CIV-L | 1/1 | 123000 |
310000 |
2.6 | |
CI cycles were prepared from D- and L-lactide under identical conditions using SnNaph2 (see Scheme 1) as catalyst.41 These CI cycles are characterized by a weight average molecular weight around 12000 g mol−1 and a low dispersity. Furthermore, their MALDI-TOF mass spectra display a large predominance of even-numbered cycles (Fig. 1A) as already reported previously.41 This predominance has the advantage that any kind of transesterification yielding odd-numbered cycles can easily be detected by mass spectroscopy. In the case of the CII–CIV cycles even- and odd-numbered cycles are fully equilibrated as exemplarily illustrated by the mass spectrum of Fig. 1B (the mass spectra of the higher cyclic polylactides are given in the ESI Fig. S1†). Furthermore, the dispersities were much higher. CII and CIII type cycles were prepared by means of the BuSnOPF catalyst (Scheme 1) in chlorobenzene solution either with a monomer concentration 0.1 mol L−1 (CII) or with a concentration of 4 mol L−1 (CIII) according to a procedure published previously.44 The CIV-L sample was prepared by means of SnBiph in bulk at 170 °C (2 h) as described in a recent publication.47 All cyclic samples were precipitated from dichloromethane solutions into ligroin to get rid of most of the catalyst. In the case of CIV-L a Mark–Houwink–Sakurada plot was measured with triple detection in comparison with a linear commercial poly(L-lactide) to make sure that this sample still consists of cycles in the high molar mass range (>100000 g mol−1) after dissolution and precipitation.
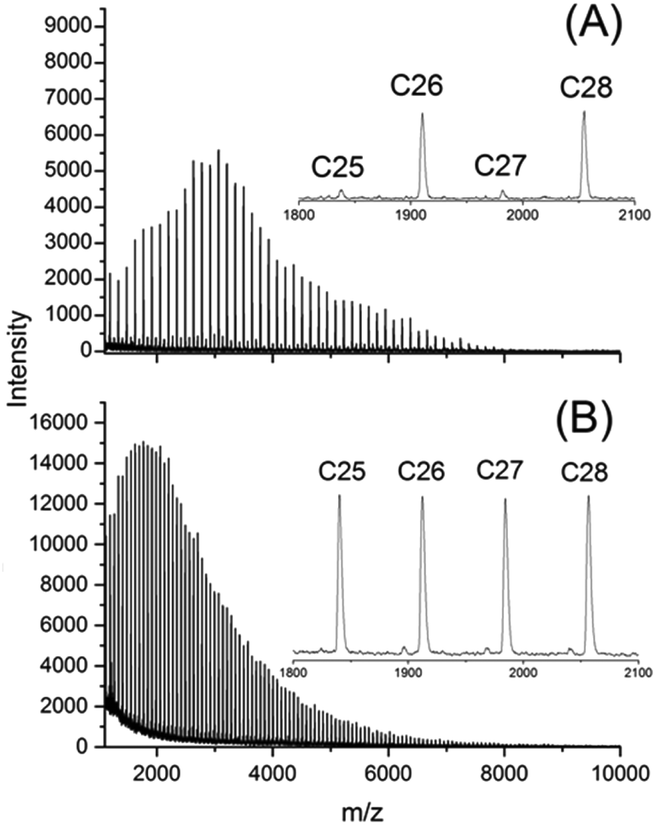 | ||
| Fig. 1 MALDI-TOF mass spectra of: (A) CI-D, (B) CIII-D. | ||
Two species of linear polylactides were synthesized using SnOct2 as catalyst and ethanol as cocatalyst which played in fact the role of an initiator (Table 2). The OH end groups were acetylated with acetic anhydride/pyridine to avoid transesterification with the cycles during annealing at high temperatures. As expected from previous publications, low dispersities and a predominance of even-numbered chains were obtained as demonstrated in Fig. 2.48,49
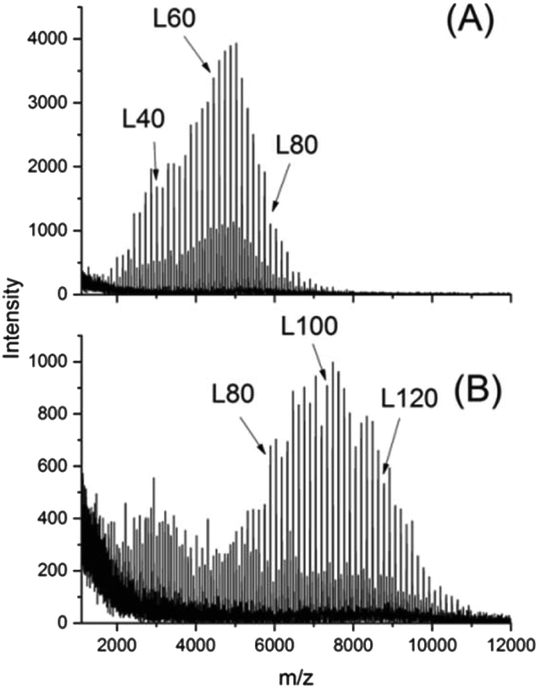 | ||
| Fig. 2 MALDI-TOF mass spectra of: (A) LI-L-Ac, (B) LII-L-Ac. | ||
Since the hydrodynamic volume of linear (but not of cyclic) polylactides is higher than that of polystyrene, polystyrene calibrated SEC measurements overestimate the real molecular weights by approximately 50%. When the measured number average molecular weights (Mn's) were multiplied with the correction factor 0.68, they were quite similar to those of the CI cycles. The three linear polylactides were also precipitated into ligroin prior to use.
Preparation of stereocomplexes
For the preparation of StCs from the enantiomeric chains four methods were reported in the literature. (1) Separate solutions of poly(D-lactide) and poly(L-lactide) in dichloromethane were mixed cast on a glass plate (e.g. Petri dish) and dried at room temperature over a period of 1 week. This procedure avoids risks with racemization or transesterification but does not guarantee 100% conversion and the isolated product may contain unreacted enantiomeric chains. (2) A combined solution of the enantiomers was precipitated into methanol. This is again a mild process, but it has two shortcomings. The polylactide chains must find each other and crystallize within seconds. Moreover, Ikada and co-workers have reported that in the case of high molar mass polylactides the conversion is incomplete, and crystallites of the neat enantiomers were detectable in addition to those of the StCs.3 When cyclic polylactides are used, this approach has the additional disadvantage that methanol slowly cleaves ester bonds (mainly during drying of the precipitates) and in the case of cycles one ester cleavage per ring suffices to change the architecture from cyclic to linear. (3) Solutions of the enantiomeric chains in acetonitrile were mixed and annealed at 80 °C for many days, whereupon the STCs crystallize from the reaction mixture. This mild process has the advantage that contamination of the StCs with unreacted parent polylactide chains is avoided. (4) The enantiomeric polylactides were mixed in the melt above 230 °C. This approach is not applicable to high molar mas polylactides (e.g. CIV) in small scale experiments, because perfect blending of the extremely viscous melts is not feasible. Furthermore, this approach runs a high risk of beginning epimerization and transesterification.The approach used in the present work resembles the “acetonitrile method”. Solutions of the enantiomeric polylactides in dichloromethane were mixed, slowly evaporated by heating and the dichloromethane was substituted by portion-wise addition of toluene. In this way the solubility of polylactides in the reaction mixture steadily decreased over a period of approx. 2.5 h. The StCs precipitated slowly from the reaction mixture in the form of crystalline powders, when the molecular weights of the starting materials were low (CI, CI, LI, LII), or in the form of gel particles, when the molecular weights were high (CIII and CIV). Washing of the virgin reaction products with warm dichloromethane removed unreacted enantiomers, if present at all. As illustrated by the data listed in Tables 3 and 4, all combinations gave quite similar yields what indicates that this procedure favoured formation of StCs from any kind of starting materials. The yields together with the DSC measurements also demonstrate that the high molecular weight of CIV-L is no hindrance for an efficient formation of StCs.
| Exp. no | Comp. | Yield (%) | T m1 (°C) | T m2 (°C) | ΔH (J g−1) | Cryst (%) DSCa | Cryst (%) WAXS |
|---|---|---|---|---|---|---|---|
| a Based on a ΔHm max of −142 J g−1 (ref. 8). b Based on 4 measurements with repeated calibration. | |||||||
| 1 | CI-D + CI-L | 77 | 202.5 | 231.5 | 88.8 | 62 | 62 |
| 2 | CI-D + CII-L | 71 | 206.5 | 229–231 | 87.2 | 61 | 59 |
| 3 | CID + CIII-L | 78 | 209.0 | 228.0–228.5 | 78.5–80.0b | 55–56 | 58 |
| 4 | CI-D + CIV-L | 69 | 218.5 | 226.0–227.0 | 72.8–75.0b | 51–53 | — |
| 5 | CIII-D + CIII-L | 72 | Shoulder | 228–229 | 72.7 | 51 | 56 |
| 6 | CIIID + CIV-L | 50 | — | 228.0 | 60.7–62.5b | 43–45 | 46 |
| Exp. no. | Comp. | Yield (%) | T m1 (°C) | T m (°C) | ΔHm (J g−1) | Cryst (%) DSCa | Cryst (%) WAXS |
|---|---|---|---|---|---|---|---|
| a Based on a ΔHm max of −142 J g−1 (s. ref. 8). b Based on four measurements with repeated calibration. | |||||||
| 1 | LI-D-Ac + LI-L-Ac | 76 | — | 233, 0–233.5 | 109.0 | 76–77b | 70 |
| 2 | CI-D + LI-L-Ac | 78 | 204.5 | 229–230 | 101.6 | 72 | 64 |
| 3 | CIII-D + LII-L-Ac | 72 | 212.0 | 230–233 | 72.5 | 50 | 59 |
DSC measurements of virgin stereocomplexes
The standard heating rate of DSC measurements used in almost all previous studies of numerous research groups is 10 K min−1 because slower heating may cause thermal degradation. The DSC traces of almost all stereocomplexes studied in this work have three important properties in common. First, no glass transition step was observable indicating a high degree of crystallinity (confirmed by WAXS measurements, see below and ESI Fig. S3†). Second, there is no melting endotherm below 195 °C what means that crystallites of neat enantiomers (stereohomopolymers) are absent. Fig. 3 and 4 exemplary illustrate these features. Third, with one exception, the minimum of the melting endotherm falls into the narrow temperature range of 228.2–231.2 °C (Tm2 in Tables 2 and 3). This is the maximum melting temperature reported by numerous research groups for StCs of linear polylactides. For the CIII-D + LII-L-Ac combination even a Tm2 around 233 °C was observed (Table 4). Hence, these DSC measurements indicate, that cyclic polylactides can form stereocomplexes with each other or with linear counterparts the crystallites of which possess the same perfection as those of linear polylactides. The cyclic topology is not a significant disadvantage from this point of view. Only combinations of very high molecular weights are unfavourable for the kinetic course of the StC formation and thus, for the crystallinity at limited reaction time (s. CI-D + CIV-L and CIII-D + CIII-L, Table 3).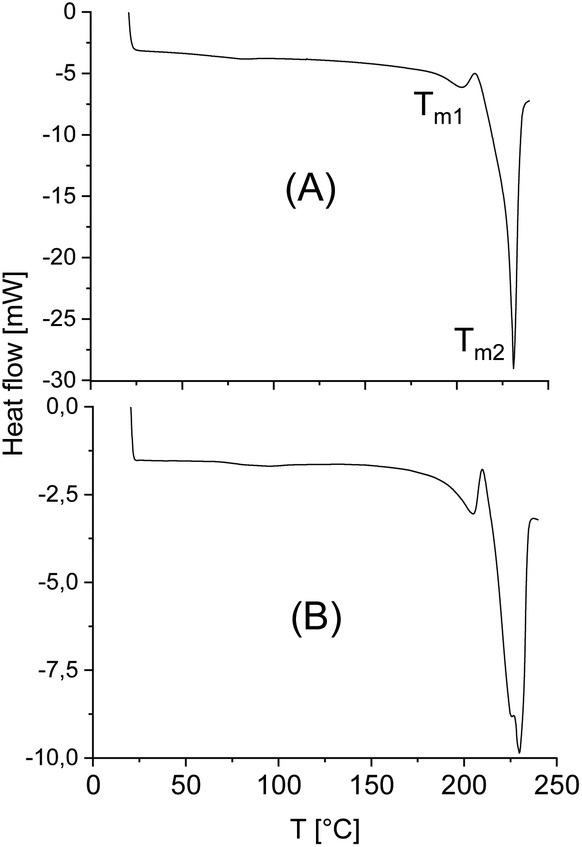 | ||
| Fig. 3 DSC curves of the virgin stereocomplexes: (A) CI-D + CI-L; (B) LI-L-Ac. | ||
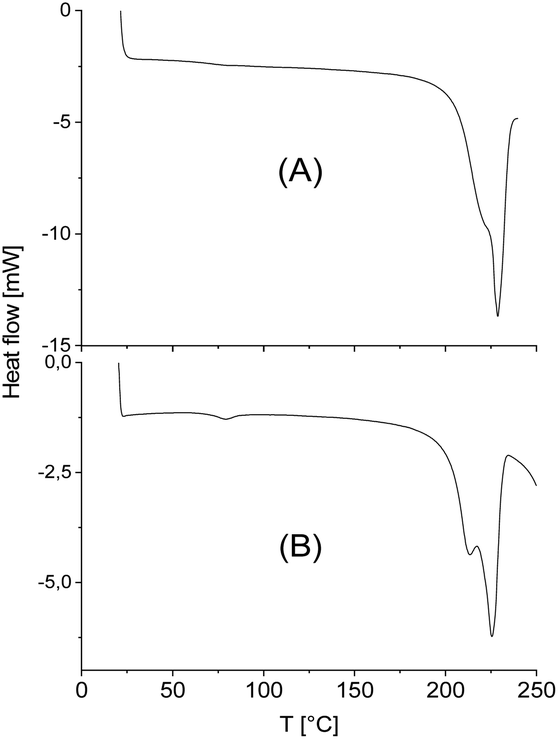 | ||
| Fig. 4 DSC curves of the virgin stereocomplexes: (A) CIII-D + CIII-L; (B) CI-D + CIV-L. | ||
However, with exceptions of “CID + CIVL” (s. Fig. 1B), all DSC curves also displayed a weak endotherm between 200 and 220 °C (see Fig. 3 and 4B and Tm1 listed in Tables 3 and 4) which was also observed by other authors for StCs of linear polylactides. Furthermore, a lower weak melting endotherm accompanying the main endotherm is also known from numerous other polymers and usually interpreted as result of a small fraction of smaller and less perfect crystallites. If this interpretation is correct, Tm1 should vanishes upon short annealing at that temperature. Tsuji et al. were the first who studied this phenomenon in the case of a linear polylactide StC and came to the same conclusion.8 The annealing experiments performed in this work are discussed below. Characteristic for StCs of the high molar mass polylactides CIII and CIV are relatively broad endotherms as illustrated by Fig. 4.
The melting enthalpies fell into the range from −70 to −90 J g−1 for StCs exclusively consisting of cycles but reached a value of −101.6 J g−1 when CI-D was combined with its linear counterpart (Table 4). The highest ΔHm was found for the StC prepared from two linear, low molar mass polylactides (Table 4). Therefore, the melting enthalpies indicate two trends. First, ΔHm increases with decreasing molecular weight of the components. Second, linear chains are more favourable for high ΔHm's than cycles. These trends are reasonable, when the StCs are prepared under identical conditions, because long chains and the cyclic topology need more time to achieve optimum conformations for StC formation and crystallization. This conclusion is supported by the annealing experiments discussed below.
The ΔHm's may, in principle, be used to calculate crystallinities, but for such calculations a comparison with a reference value for 100% crystallinity is necessary.
At this point it should be mentioned that despite numerous publications, crystallinities of StCs derived from linear polylactides have rarely been reported. Only one reference value has been found by the authors, namely −142 J g−1 used by Tsuji et al. for the calculation of crystallinity of a sample annealed at 216 °C for 10 min.8 Those authors cited a paper of Loomis et al. (ref. 21 in this work) as source of this value, which is an abstract of a talk presented at the ACS meeting in 1990. It does not mention melting enthalpies and how they were determined. In 2005 Sarasua et al.15 reported crystallinities for numerous StC samples (after variation of the annealing conditions) calculated from DSC measurements with the same ΔHm max. Surprisingly, also, those authors cited the “Loomis paper” as source.15,24 Hence, origin, and margin of error of this ΔHm max are uncertain. Usually, a ΔHm max is calculated by extrapolation of ΔHm values of samples the crystallinity of which is determined by WAXS measurements. These WAXS measurements have a margin of error around 5% and thus, the extrapolated ΔHm max is also afflicted with a similar margin of error.
Nonetheless, crystallinities calculated with a ΔHm max of −142 J g−1 were listed in Tables 3–5 for comparison with the data of Tsuji et al.8 and Sarasua et al.15 The highest value found by Tsuji was 70% crystallinity after annealing at 216 °C for 10 min and by Sarasua, who found a value of 61.5% after annealing at 190 °C for 24 h. Those values agree well with the highest crystallinities calculated analogously in this work (see Tables 3 and 5).
| Comp. | Annealing at 160 °C/1 d | Annealing at 205 °C/1 h | |||||
|---|---|---|---|---|---|---|---|
| T m (°C) | ΔHm (J g−1) | Cryst (%) DSCa | Cryst (%) WAXS | T m (°C) | ΔHm (J g−1) | Cryst (%) DSCa | |
| a Based on a ΔHm max of −142 J g−1 (s. ref. 8). | |||||||
| CI-D + CI-L | 214.0 | 96.5 | 68 | 73 | 220.5 | 80.8 | 57 |
| CI-D + CII-L | 212.0 | 107.8 | 76 | 81 | 217.2 | 86.6 | 61 |
| CI-D + CIII-L | 212.0 | 97.5 | 69 | 69 | 214.5 | 83.6 | 59 |
| CID + CIVL | 230.5 | 85.5 | 60 | — | 225.5 | 85.7 | 60 |
| CI-D + LI-L-Ac | 216.5 | 104.0 | 74 | 69 | 216 | 83.5 | 59 |
| CIII-D + CIII-L | 217.5 | 103.0 | 73 | 69 | 232.0 | 95.5 | 67 |
| CIII-D + LII-L-Ac | 215.5 | 103.5 | 73 | 74 | 220.0 | 97.5 | 69 |
WAXS of virgin stereocomplexes
The WAXS powder patterns displayed all those reflections known for the crystal lattice of linear StCs (see ESI Fig. S2†). Reflections of crystalline stereohomopolymers were not detectable, so that the WAXS measurements agree with the DSC traces in that crystallites of unreacted stereohomopolymers were absent. Hence, it may be concluded that the experimental procedure used in this work allows for the preparation of neat StCs.The WAXS powder patterns of most all samples displayed a rather flat baseline indicating crystallinities above 50% (Fig. S3†). Only the combination of two high molar mass cycles (CIII-D + CIV-L) gave a curved baseline yielding a crystallinity below 50% (Table 3). The trends that can be derived from these WAXS crystallinities are the same as those extracted from the DSC measurements. High crystallinities are favoured by lower molecular weights and by the linear topology. In the case of the three StCs prepared by combination of cyclic and linear polylactides the crystallinities derived from DSC measurements vary over a broader range than those calculated from WAXS patterns. The reproducibility of both, DSC and WAXS measurements has been checked and an explanation for this difference cannot forwarded at this time, but it does not affect the conclusions and trends presented above.
SAXS measurements show only diffuse scattering and a lamellar long period signal cannot be detected (see ESI Fig. S3†). The lack of SAXS reflections may be explained by diffuse boundaries between the crystal lamellas and the interlamellar amorphous regions, and a short distance between the crystallites.
The effect of annealing
Annealing at 160 °C for 1 d was performed, because it was learned from numerous polymerization experiments in previous studies that this temperature does not involve racemization or thermal degradation for period of 1 d, so that the physical effect of annealing may be studied without interference of chemical reactions. Annealing at 205 °C for 1 h has firstly been performed to study the influence on Tm1. One similar experiment has been published by Tsuji et al.,7 who annealed a StC of two linear polylactides for 10 min at 216 °C and observed the expected disappearance of Tm1. Secondly, it should be found out, if random transesterification with formation of D-L/L-D sequences occurs. Randomization of the stereosequence has the consequence, that the crystallinity considerably decreases and finally even disappears.The annealing at 160 °C had the expected consequence that the degree of crystallinity increased as demonstrated by the ΔHm values listed in Table 5 compared with those listed in Table 3. This increase is particularly pronounced, when the initial crystallinity was relatively low (according to DSC), as it is typical for StCs including large cycles such as CIII-L and CIV-L. The WAXS data confirm this trend, although the absolute values were approximately 10% higher. At this point it is of interest to compare the melting enthalpies of the cyclic stereocomplexes (−85.5 up to −107.8 J g−1, Table 5) with the highest values reported for annealed StCs of linear polylactides. Tsuji found −98 and −102 J g−1 for samples annealed at 160 °C for 1 h or 216 °C for 10 min).8 Sarasua obtained a ΔHm of −87.3 J g−1 for a sample annealed at 190 °C for 24 h.15 These data demonstrate, that after annealing cyclic StCs can reach the same level of crystallinity as the best samples of linear StCs, even when high molar mass cycles are involved. This result needs to be underlined, because Tsuji et al. concluded from mixing linear polylactides of different molecular weights, that a large extent of StC formation requires Mw's below 4 × 104 g mol−1, and around 4 × 105 g mol−1 StC formation should become impossible.3 Sarasua later concluded: “Stereocomplexation hardly develops when mixing high molecular stereoisomers, a result related to the smaller overall mobility of high molecular weight chains compared to that of lower molecular weight ones”.15
The DSC traces of the six samples selected for the annealing experiments at 205 °C (see Table 5, last two columns) gave the following results. First, no glass transition step was detectable indicating that no large fraction of amorphous material was formed. Second, in all cases, where a weak melting endotherm below 220 °C (i.e. Tm1) was detected in the virgin stereocomplexes, this melting endotherm had vanished. Both observations are proved by the DSC curve of Fig. 5. Third, the melting enthalpies were higher than those of the virgin StCs, but they were lower than those obtained at 160 °C and thus, suggest that these results were either influenced by beginning random transesterification and/or thermal degradation. Hence, both, the DSC curves and the WAXS powder patterns carried the same message, namely absence of significant transesterification reactions generating a randomization of the homosteric D- and L-chains.
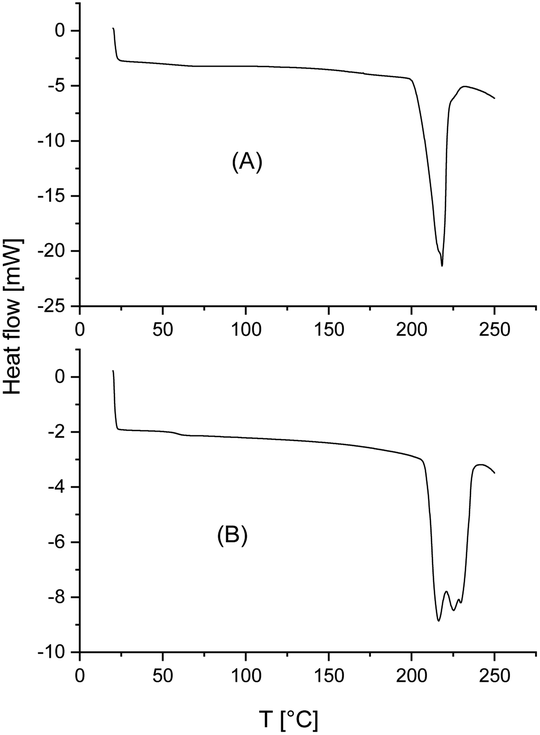 | ||
| Fig. 5 DSC heating curves recorded after annealing at 205 °C/1 h: (A) CI-D + CII-L, (B) CID + CIVL. | ||
To confirm this conclusion by a more precise method, both, 1H and 13C NMR spectra of two StCs (CI-D + CII-L and CIII-D + CIII-L) annealed at 160 or 205 °C were recorded and compared with those of the virgin StCs (see ESI Fig. S4†). No difference was found between annealed and virgin samples. From polylactides it is well known, that the CO and CH signals are sensitive to different stereosequences, so that signals of different D-L/L-D containing triads or tetrads are detectable.50,51 Such signals were not observable in the spectra of the annealed samples (see ESI Fig. S5†). In this spectrum seven of the eight possible D-L/L-D tetrads are clearly observable, whereas only the isotactic tetrad shows up in the spectra of the annealed stereocomplexes. These NMR spectra were recorded with a 600 MHz NMR spectrometer and the 13C NMR scans were accumulated over a period of 12 h, so that a S/N ratio >150/1 was achieved for the CO signal of the stereocomplexes. Therefore, it may reliably be concluded, that the percentage of D-L/L-D diads (if any) did certainly not exceed 5%, a value which might be explained by transesterification in the amorphous phase.
This information is important, because the MALDI TOF mass spectrum of the CI-D + CI-L StC annealed at 205 °C told a somewhat different story (Fig. 6). As demonstrated by comparison of Fig. 6 with Fig. 1A (representing the starting materials), the predominance of the even-numbered cycles was still maintained but the fraction of odd-numbered cycles had increased from approx. 5% to 30%. Considering, that the 13NMR spectra proved almost total absence of random transesterification, the mass spectrum of Fig. 6 indicates that a second unusual transesterification mechanism was operating enabling selective ring–ring equilibration. This conclusion is supported by the fact that the intensity distribution of the “odd-peaks” parallels that of the “even peaks” and no broadening of the peak pattern has occurred as it may be expected for random transesterification. A satisfactory formulation of this unusual ring–ring equilibration cannot be forwarded at this time, but a detailed study of this phenomenon and other transesterification reactions in the solid state of cyclic polylactides is in progress and will be published in the near future.
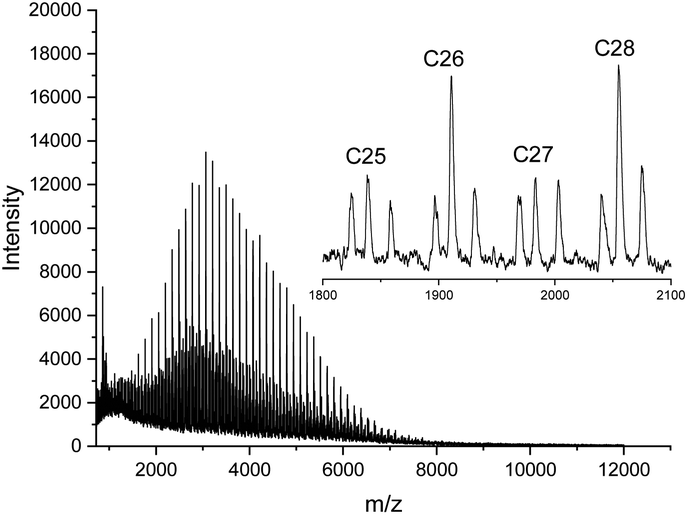 | ||
| Fig. 6 MALDI TOF mass spectrum of the stereocomplex CI-D + CI-L after annealing at 205 °C for 1 h. | ||
The mass spectra of CID + CIL annealed at 160 and 205 °C also displayed peaks of linear chains resulting from thermal degradation. Since the crystallinity increased upon annealing these degradation reactions must be limited to the disordered phase between the crystallites and to the surface of the crystallites. Hence, these preliminary results demonstrate that the combination of NMR spectroscopy and MALDI TOF mass spectrometry is a powerful tool to study transesterification and other chemical reactions in the solid state of cyclic (and linear) polylactides, provided that these polylactides contain a large excess of either even or odd-numbered species.
The degradation reactions indicated by the mass spectra were confirmed by dynamic thermogravimetric measurements with a heating rate of 5 K min−1. In the case of CI-D + CI-L the loss of weight begins around 200 °C and reaches 5% at 222 °C (s. ESI Fig. S6†). This unexpectedly low thermal stability most likely results from the presence of the catalyst SnNaph2 which was used in large amounts for the preparation of the parent stereohomocycles and which is difficult to remove completely. When high molar mass cycles are prepared with small quantities of catalysts, the thermostability is considerably higher.
For example, CI-D + CIV-L shows 5% loss of weight at 242 °C (see ESI Fig. S7†) and CIV-L alone at 262 °C (see ESI Fig. S8†). This is the highest thermostability the authors have found for a polylactide so far, regardless if cyclic or linear. A detailed study of thermostabilities of cyclic and linear polylactides and their stereocomplexes is in progress and will presented in a future publication.
Conclusions
The results of this work show, that the method used for the preparation of StCs yields crystalline StCs bare of homostereocopolymers. Furthermore, as indicated by the Tm values, all types of cycles studied in this work were capable to form crystallites of StCs that have the same perfection as those derived from linear polylactides. However, at limited reaction time the extent of crystallinity decreases with increasing molecular weight of the cycles, because large cycles need more time to acquire the optimum conformation needed for formation of StCs and their crystallization. This kinetic short-coming disappears upon annealing, so that crystallinities above 70% can be reached even with StCs derived from high molar mass cycles. Annealing at 160 °C for 1 d or at 205 °C for 1 h revealed that significant random transesterification with formation of D-L/L-D sequences does not occur, whereupon special even-odd ring-ring equilibration may proceed. Based on this observation and other unpublished results a more detailed investigation on ring–ring equilibration and other transesterification reactions occurring in the solid state of cyclic polylactides and their StCs is now in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Thyssen-Uhde AG (Berlin) for having kindly supplied L- and D-lactide. Furthermore, we thank Prof. H. U. Moritz (TMC, Univ. Hamburg) for financial support and the Federal Institute for Materials Research and Testing (BAM) for instrumental support.Notes and references
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS
.
- H. Tsuji, S. H. Hyon and Y. Ikada, Macromolecules, 1992, 25, 2940–2946 CrossRef CAS
- H. Tsuji, S. H. Hyon and Y. Ikada, Macromolecules, 1991, 24, 5657–5662 CrossRef CAS
- H. Tsuji, S. H. Hyon and Y. Ikada, Macromolecules, 1991, 24, 5651–5656 CrossRef CAS
- H. Tsuji, F. Horii, S. H. Hyon and Y. Ikada, Macromolecules, 1991, 24, 2719–2724 CrossRef CAS
- H. Tsuji and Y. Ikada, Macromolecules, 1993, 26, 6918–6926 CrossRef CAS
- H. Tsuji and Y. Ikada, Macromolecules, 1992, 25, 5719–5723 CrossRef CAS
- H. Tsuji, F. Horii, M. Nakagawa, Y. Ikada, H. Odani and R. Kitamaru, Macromolecules, 1992, 25, 4114–4118 CrossRef CAS
- T. Okihara, M. Tsuji, A. Kawaguchi, K. Katayama, H. Tsuji, S. H. Hyon and Y. Ikada, J. Macromol. Sci., Part B: Phys., 1991, 30, 119–140 CrossRef CAS
- S. Lee, M. Kimoto, M. Tanaka, H. Tsuji and T. Nishino, Polymer, 2018, 138, 124–131 CrossRef CAS
- H. Tsuji and Y. Arakawa, Polym. Chem., 2018, 9, 2446–2457 RSC
- H. Tsuji, K. Kikkawa, R. Ozawa and Y. Arakawa, CrystEngComm, 2019, 21, 3158–3169 RSC
- H. Tsuji, Y. Arakawa and N. Matsumura, Polym. Bull., 2019, 76, 1199–1216 CrossRef CAS
- H. Tsuji, M. Yamasaki and Y. Arakawa, ACS Appl. Polym. Mater., 2019, 1, 1476–1484 CrossRef CAS
- J. R. Sarasua, N. L. Rodriguez, A. L. Arraiza and E. Meaurio, Macromolecules, 2005, 38, 8362–8371 CrossRef CAS
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed
- K. Fukushima and Y. Kimura, Polym. Int., 2006, 55, 626–642 CrossRef CAS
- M. Kakuta, M. Hirata and Y. Kimura, Polym. Rev., 2009, 49, 107–140 CrossRef CAS
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS PubMed
- H. W. Bai, S. H. Deng, D. Y. Bai, Q. Zhang and Q. Fu, Macromol. Rapid Commun., 2017, 38, 1700454 CrossRef PubMed
- G. L. Loomis, J. R. Murdoch and K. H. Gardner, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1990, 31(2), 55 Search PubMed
- S. Brochu, R. E. Prudhomme, I. Barakat and R. Jerome, Macromolecules, 1995, 28, 5230–5239 CrossRef CAS
- L. Cartier, T. Okihara and B. Lotz, Macromolecules, 1997, 30, 6313–6322 CrossRef CAS
- J. R. Sarasua, A. L. Arraiza, P. Balerdi and I. Maiza, J. Mater. Sci., 2005, 40, 1855–1862 CrossRef CAS
- N. Yui, P. J. Dijkstra and J. Feijen, Makromol. Chem., 1990, 191, 481–488 CrossRef CAS
- N. Spassky, M. Wisniewski, C. Pluta and A. LeBorgne, Macromol.
Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS
- J. R. Sarasua, R. E. Prud'homme, M. Wisniewski, A. Le Borgne and N. Spassky, Macromolecules, 1998, 31, 3895–3905 CrossRef CAS
- C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552–1553 CrossRef CAS
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS PubMed
- Z. Y. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed
- K. Majerska and A. Duda, J. Am. Chem. Soc., 2004, 126, 1026–1027 CrossRef CAS PubMed
- Z. H. Tang, X. S. Chen, X. Pang, Y. K. Yang, X. F. Zhang and X. B. Jing, Biomacromolecules, 2004, 5, 965–970 CrossRef CAS PubMed
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed
- C. X. Cai, A. Amgoune, C. W. Lehmann and J. F. Carpentier, Chem. Commun., 2004, 330–331 RSC
- K. Fukushima and Y. Kimura, Macromol. Symp., 2005, 224, 133–143 CrossRef CAS
- K. Fukushima, Y. Furuhashi, K. Sogo, S. Miura and Y. Kimura, Macromol. Biosci., 2005, 5, 21–29 CrossRef CAS PubMed
- N. Sugai, T. Yamamoto and Y. Tezuka, ACS Macro Lett., 2012, 1, 902–906 CrossRef CAS
- N. Sugai, S. Asai, Y. Tezuka and T. Yamamoto, Polym. Chem., 2015, 6, 3591–3600 RSC
- E. J. Shin, A. E. Jones and R. M. Waymouth, Macromolecules, 2012, 45, 595–598 CrossRef CAS
- S. M. Weidner and H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2730–2738 CrossRef CAS
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Macromol. Chem. Phys., 2017, 218, 17000274 CrossRef
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 109, 360–366 CrossRef CAS
- H. Kricheldorf, S. M. Weidner and F. Scheliga, Eur. Polym. J., 2019, 116, 256–264 CrossRef CAS
- G. Benecke, W. Wagermaier, C. H. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Muller-Buschbaum, M. Trebbin, S. Forster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Eur. Polym. J., 2019, 116, 256–264 CrossRef CAS
- S. Sosnowski and P. Lewinski, Polym. Chem., 2015, 6, 6292–6296 RSC
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2018, 219, 1800445 CrossRef
- H. R. Kricheldorf, C. Boettcher and K. U. Tonnes, Polymer, 1992, 33, 2817–2824 CrossRef CAS
- H. R. Kricheldorf and C. Boettcher, Macromol. Chem., 1993, 194, 1653–1664 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01236b |
| This journal is © The Royal Society of Chemistry 2019 |