
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(Boc-acryloyl hydrazide): the importance of temperature and RAFT agent degradation on its preparation†
Oliver
Creese
,
Pavan
Adoni
,
Guanlong
Su
,
Andrey
Romanyuk
 and
Paco
Fernandez-Trillo
*
and
Paco
Fernandez-Trillo
*
School of Chemistry, and Institute of Microbiology and Infection, University of Birmingham, Edgbaston, B15 2TT Birmingham, UK. E-mail: f.fernandez-trillo@bham.ac.uk
First published on 3rd October 2019
Abstract
Poly(acryloyl hydrazide) is a versatile polymer scaffold readily functionalised through post-polymerisation modification with aldehydes to yield polymers for biological applications. However, its polymerisation is affected by nucleophilic degradation of the RAFT agent that leads to early termination, an issue often overlooked in the polymerisation of primary acrylamides. Here we report the effect of temperature on the RAFT polymerisation of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1) and demonstrate that by carefully selecting this polymerisation temperature, a balance between rate of polymerisation and rate of degradation of the RAFT agent can be achieved. This way greater control over the polymerisation process is achieved, allowing the synthesis of Boc-protected poly(acryloyl hydrazide) with higher degrees of polymerisation (DP) than those obtained previously, while still maintaining low dispersities (ĐM).
Introduction
Synthetic polymers are increasingly becoming an attractive means of interfacing biological systems via multivalent binding, displaying activities orders of magnitude higher than those of their monovalent components.1–5 Thus, polymers are now widely researched for biomedical applications including as antimicrobials,6,7 as drug and gene delivery vehicles,2,8 as biological sensors,9,10 or as “smart” biomaterials with anti-fouling properties.11 Highly functional polymers developed for specific applications generally involve the use of functional monomers which either already possess the final desired functionality, or have the capability of undergoing post-polymerisation modification to introduce the desired functionality. This latter approach can greatly broaden the scope of chemical functionalities used. Post-polymerisation modification has normally relied on click chemistries,12 and has now been greatly expanded through the use of oxime13 and hydrazone chemistry,14,15 reductive amination,16 and epoxide ring opening.17A common limitation when developing synthetic polymers for biomedical applications is the need to screen large libraries of compounds which is costly and time consuming. In this regard, poly(acryloyl hydrazide) has been recently reported as a versatile platform for the synthesis and screening of polymers for biomedical applications.14,18–20 Functional polymers are obtained by simple incubation of poly(acryloyl hydrazide) with functional aldehydes, both under aqueous or organic conditions,14 and this polymer has now been applied to the development of glycan arrays,18 pH sensitive drug-delivery,21 and nucleic acid delivery.20,22,23 In our laboratories poly(acryloyl hydrazide) was prepared from Boc-protected precursor Boc-Px (Scheme 1) following deprotection with TFA.14 Reversible addition–fragmentation chain-transfer (RAFT) polymerisation of N′-(tert-butoxycarbonyl) acryloyl hydrazide (1) resulted in a small library of polymers. However, control over the polymerisation was lost with increasing conversion and degree of polymerisation, possibly as a result of degradation of the RAFT agent through intramolecular nucleophilic attack. This degradation has been reported in the RAFT polymerisation of other acrylamide derivatives,24,25 including closely related methacryloyl hydrazide,26 with better control reported when the polymerisation is carried out at low temperatures.25,27 This side-reaction is often overlooked in the polymerisation of primary and secondary acryl- and methacrylamides, and makes synthesising highly functional polymers from this type of monomers inherently challenging.28 The need for greater control over these materials is more significant when looking to understand better the nature of the structure–activity relationship throughout post-polymerisation modification and biological screening.
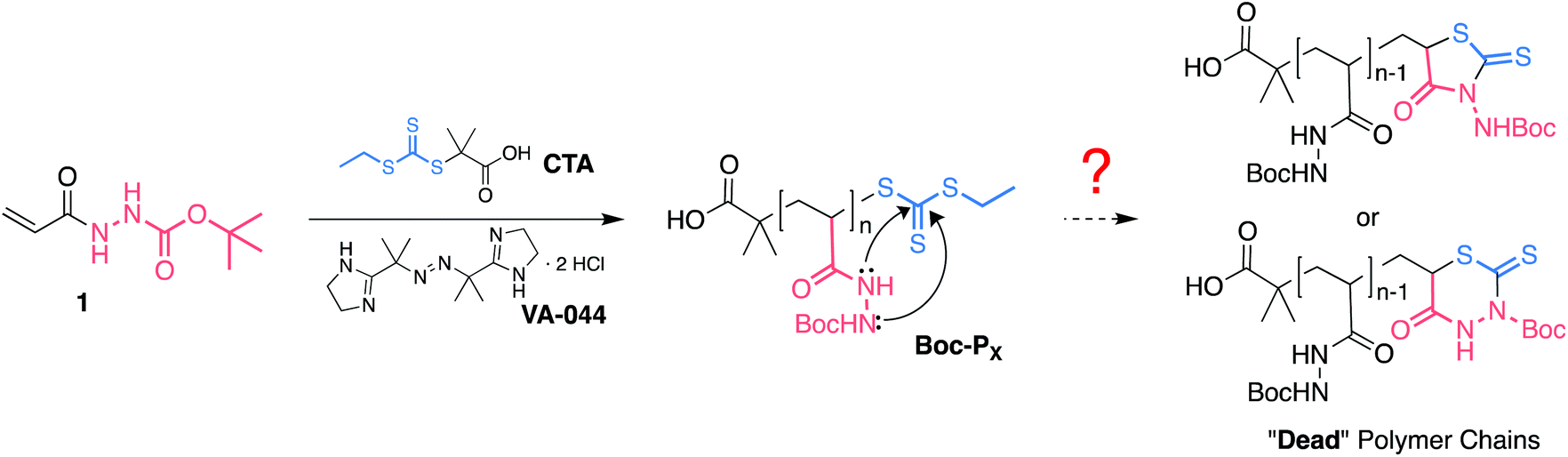 | ||
| Scheme 1 RAFT polymerisation of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1) and potential degradation by-products. | ||
Here, we report the effect of temperature and the decomposition rate of the initiator on the polymerisation of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1), as a route to optimise the preparation of poly(acryloyl hydrazide). Polymerisations were carried out using 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) as a low temperature initiator, so that the rate of generation of radicals could be readily modified as a function of temperature. Our results suggest that while increasing the temperature increases the polymerisation rate, it also speeds up RAFT degradation and thus, loss of control. Conditions have been identified for which the polymerisation “outperforms” this side reaction and polymers with good control over molecular mass and dispersities (ĐM) can be obtained. More importantly, these conditions allowed us to prepare Boc-Px with higher degrees of polymerisation and lower ĐM, not accessible with our previous conditions.14 This improved control over the polymerisation of Boc-protected poly(acryloyl hydrazide) will be of value when DP and ĐM may underpin future applications.
Experimental section
Materials
2-((Ethylthio)carbonothioyl)thio-2-methylpropanoic acid (CTA)29 and N′-(tert-butoxycarbonyl)acryloyl hydrazide (1)14,20 were synthesised according to protocols described in the literature. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) was purchased from Fluorochem and used without further purification. All other chemicals were purchased from Sigma-Aldrich®, Fisher Scientific®, VWR® or Acros®, and used without further purification. All solvents were Reagent grade or above, purchased from Sigma-Aldrich®, Fisher Scientific® or VWR®, and used without further purification. Polymethylmethacrylate standards were purchased from Agilent®.Characterisation
Nuclear Magnetic Resonance (NMR) spectra were recorded on either a Bruker Avance III 300 MHz or a Bruker Avance III 400 MHz spectrometer. Chemical shifts are reported in ppm (units) referenced to the following solvent signals: dimethylsulfoxide (DMSO)-d6 H 2.50. Gel Permeation Chromatography (GPC) was performed with a Shimadzu Prominence LC-20A fitted with a Thermo Fisher Refractomax 521 Detector and a SPD20A UV-vis Detector. Poly(N′-(tert-butoxycarbonyl)acryloyl hydrazide) (Boc-Px) was analysed using 0.05 M LiBr in dimethylformamide (DMF) at 60 °C as the eluent, and a flow rate of 1 mL min−1. The instrument was fitted with a Polymer Labs PolarGel guard column (50 × 7.5 mm, 5 μm) followed by two PLGel PL1110-6540 columns (300 × 7.5 mm, 5 μm). Molecular masses were calculated based on a standard calibration method using polymethylmethacrylate standards.RAFT polymerisation of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1)
In a typical kinetic experiment 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) (11.7 mg, 0.036 mmol), 2-ethylthiocarbonothioylthio-2-methylpropanoic-acid (CTA) (40.3 mg, 0.18 mmol) and N′-(tert-butoxycarbonyl)acryloyl hydrazide (1) (1.666 g, 8.950 mmol) were dissolved in DMSO (10.0 mL) and a 100 μL sample was taken at this stage to calculate conversion (ρ). The solution vessel was sealed with a septum, securely fastened with electrical tape to maintain the seal, and degassed by bubbling with argon for 25 minutes. Using a cannula, 1 mL of the solution was transferred to sealed glass vials containing stirrer bars, each degassed for 5 minutes. Vials were then left to react at a pre-set temperature (30–150 °C) for the required amount of time. The reaction was stopped by allowing the tube to cool using a water bath and exposing it to air. 100 μL aliquots of each timepoint were taken at this stage to calculate conversion (ρ) and for GPC analysis. NMR and GPC analysis of each timepoint was carried out from the crude mixture. The natural logarithm of the inverse of the fractional concentration of monomer – ln(M0/Mt) – was plotted against time, and the data fitted using GraphPad Prism version 6.0 for Mac Os X, GraphPad Software, La Jolla California USA, http://www.graphpad.com. The in-built segmental line regression was used to fit the data to two intersecting lines. This model was used to identify when a change in the polymerisation kinetics was observed (tdead).Results and discussion
As reported, our initial efforts to optimise the polymerisation of Boc-protected acryloyl hydrazide 1 focused on reducing the temperature of the polymerisation.14 RAFT polymerisation of acrylamides and methacrylamides often suffers from cleavage of the RAFT agent through intramolecular addition–elimination of the weakly nucleophilic amides to the trithiocarbonate group (Scheme 1).25 Under our previously reported conditions for the polymerisation of 1, a change in the rate of polymerisation was observed with increasing conversion (Fig. S1A†) which we associated with this degradation of the terminal trithiocarbonate in the growing chain. It has been proposed that reducing the polymerisation temperature would significantly reduce the rate of this side reaction.25 Thus, optimisation of the polymerisation was at that time carried out under the same conditions but using initiators with different 10 hours half-life decomposition temperatures (t10) (Fig. S1†). This way, the rate of formation of radicals was kept as similar as possible for all polymerisations while reducing the temperatures to 50 °C (V-65) or 44 °C (VA-044). Despite the use of lower temperatures, in all cases, a change in the kinetics of the polymerisation was observed, although this change was not as obvious for the polymerisations performed at 44 °C (Fig. S1A,† right). To identify when this change in rate of polymerisation was occurring, the natural logarithm of the inverse of the fractional concentration of monomer – ln(M0/Mt) – was plotted against time, and the data fitted to a segmental line regression. This function fits the data to two different lines, before and after a breakpoint. In our case, we termed the breakpoint tdead because we think that after this point, side reactions have a predominant effect on the kinetics of the polymerisation resulting in an increasing number of dead polymer chains. This change in kinetics was reflected on the relatively high dispersity in molecular mass (ĐM = 1.38–1.95) obtained for the polymers prepared under these conditions.14 Overall, no clear benefit from reducing the temperature was observed, with a tdead of approximately 4 and 4.5 hours for polymerisations at 50 °C and 70 °C respectively. Interestingly, tdead for the polymerisation performed at 44 °C was observed at approximately 2.5 h, which would suggest degradation was occurring faster at this temperature. This was not expected and may suggest that other mechanisms beyond the simple degradation of the RAFT agent may be at play. For instance, polymerisation decay can also be caused by diminishing initiator efficiency at high monomer conversions, which has been observed for some azo-initiators.30,31 However, this factor normally becomes significant at much higher conversions than the ones we reported.Attempts to perform the polymerisation at an even lower temperature (30 °C) using VA-044 as the source of radicals resulted in a very long induction period followed by a short period of linear increase of the fractional concentration of monomer until a change in kinetics was again evident (Fig. S2†). The maximum conversion in this case was 50% – ln(M0/Mt) = 0.83, worse than that observed for the polymerisations performed at higher temperatures.
In order to determine if degradation of the RAFT agent was indeed possible at low temperatures, we attempted to synthesise a small molecule analogue which mimicked an n = 1 polymer (Scheme S1†). To this end, 2-bromopropionic acid (2) was reacted with tert-butyl carbazate, and the resulting bromine derivative 3 reacted under standard conditions for the formation of the RAFT agent. 1H NMR analysis of this reaction revealed a very complex mixture, where only traces of something that could resemble trithiocarbonate 4 could be identified (Fig. S4†). This observation was in line with our previous results, and suggested that hydrazide containing trithiocarbonates such as 4 were very amenable to intramolecular nucleophilic attack. Attempts to isolate this trithiocarbonate 4 were unsuccessful, with the main isolated product of this reaction being tentatively assigned to a mixture of the 5- and 6-membered rings in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio (Fig. S5†).
:4 ratio (Fig. S5†).
Seeing how lowering the temperature had no beneficial effect on the kinetics of the polymerisation of 1, and a change in kinetics was still observed, we decided to explore the use of “Ultra-Fast” polymerisation conditions in an attempt to outrun the side reaction.32–34 Our hypothesis was that by using a low temperature initiator such as VA-044 at a significantly higher temperature (e.g. 100 °C) than the reported t10 (44 °C), an increase in the concentration of radicals in solution would be achieved. This way, the concentration of propagating radicals would be higher with a greater number of chains growing at the same time, resulting in the synthesis of polymers with better control over the molecular mass and ĐM. This methodology is particularly suitable for fast-propagating monomers such as acrylamides, and since the rate of polymerisation is directly proportional to the concentration of these propagating radicals (and the monomer concentration, Rp = kp[M][P˙]), we postulated that running the polymerisation under these conditions could outperform the side reaction observed under standard RAFT polymerisation conditions. In a first attempt, the polymerisation conditions previously reported by us for the polymerisation of 1 (Fig. S1†)14 were modified so that the initiator used was VA-044 and the polymerisation temperature was 100 °C. A shorter polymer was targeted this time and, as expected, the polymerisation was very fast, reaching up to 70% conversion in less than five minutes (Fig. 1A, CTA:VA-044 5:1 ●). The change in reaction rate could not be suppressed and was again evident, with a tdead of approximately 4.5 min. Before tdead, the polymerisation retained the features of a controlled polymerisation, with the molecular mass of the polymer directly proportional to the conversion and, comparable dispersities (Fig. 1B, left) to those observed with our previous conditions.14
 | ||
| Fig. 1 (A) Plot of conversion (ρ) vs. time and (B) measured number average molecular mass (Mn) vs. conversion (●) and dispersity in molecular mass (ĐM) vs. conversion (○), for polymerisations of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1) performed with different CTA:VA-044 ratios. Conditions: [M] = 0.9 M, [M]/[CTA] = 50/1. Mn and ĐM calculated by GPC using 0.05 M LiBr in dimethylformamide (DMF) at 60 °C. | ||
These results were promising and we therefore explored decreasing the concentration of initiator in our polymerisations, in an attempt to suppress termination, increase the number of chains growing from the RAFT agent and thus optimising the dispersities. However, while dispersities were decreased, reducing the concentration of initiator in these polymerisations resulted in slower reactions, with no effect observed in tdead (Fig. 1A). As a result, the maximum conversion obtained when the CTA:VA-044 ratio was increased to 10:1 or 15:1 (40% and 24% conversion respectively) was lower than in the previous case (70%).
We decided next to run the polymerisations at 150 °C, in an attempt to further increase the concentration of radicals during early stages of polymerisation, and thus the rate of propagation. However, these conditions not only resulted in lower conversions (Fig. S9†) but a colour change of the reaction mixture from yellow to dark brown, suggesting that thermal decomposition of the trithiocarbonate group was ocurring.35 Thermal decomposition of the RAFT agent was confirmed via1H NMR where signals consistent with the β-elimination of the trithiocarbonate could be observed (Fig. S10†).35,36
Having identified conditions to run the polymerisation of 1 at 100 °C, which resulted in similar conversions and dispersities to those previously reported, we decided to explore the use of these conditions to prepare polymers of higher DP (Fig. 2), which were harder to control using our previously reported method.14 Three different DPs were targeted (i.e. [1]/[CTA] = 50, 100 and 150), by maintaining the concentration of 1 and reducing the amount of RAFT agent and initiator used. As expected, this resulted in slower polymerisations, while tdead still remained at around 4.5 min (Fig. 2A). As a consequence, polymerisations targeting 100 and 150 monomer units only reached low conversions (∼40% and 30% respectively). In any case, control over the molecular mass of the polymer was still observed during the first stages of the polymerisation, with the average molecular mass (Mn) increasing linearly with time until the change in polymerisation rate was evident (tdead) (Fig. 2B). A clear shift towards lower retention time was observed in the gel permeation chromatograms when higher DPs were targeted, suggesting that, at least during the initial phase of the reaction, the polymerisation was maintaining features of a controlled radical polymerisation. ĐM remained similar across the three targeted molecular masses which demonstrates an improvement compared to our previous conditions where ĐM increased with increasing targeted DP.
 | ||
| Fig. 2 (A) Plot of conversion (ρ) vs. time, (B) fractional concentration of monomer ln(M0/Mt) vs. time, and (C) measured number average molecular mass (Mn) vs. conversion (ρ) (top) and dispersity in molecular mass (ĐM) vs. conversion (ρ) (bottom), for polymerisations of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1) performed at 100 °C with different 1:CTA ratios. (D) GPC chromatograms of the resulting polymers at the highest conversion obtained. Conditions: [M] = 0.9 M, [CTA]/[VA-044] = 5/1. Mn and ĐM calculated by GPC using 0.05 M LiBr in dimethylformamide (DMF) at 60 °C. | ||
At this point, our results suggested that a compromise could be obtained between increasing the rate of propagation by increasing the polymerisation temperature, and delaying tdead by reducing the polymerisation temperature. Therefore, we investigated polymerisations at intermediate temperatures (Fig. 3). While a change in polymerisation rate was still evident for the new temperatures investigated, higher conversions could be achieved for the polymerisation performed at 65 °C (90%) while the next highest conversions at 80 °C and 50 °C were 77% and 80% respectively (Fig. 3B, ●). Temperature had a significant effect on the time at which a change in polymerisation rate was evident (tdead), with this inflection point happening sooner as the temperature was increased (Fig. 3B, ○).
 | ||
| Fig. 3 (A) Plot of fractional concentration of monomer ln(M0/Mt) vs. time for polymerisations of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1) performed at different temperatures. (B) Effect of temperature on the time at which deviation from linearity for the plot of ln[M]0/[M]tvs. time is observed (tdead) (○), and the fractional concentration of monomer ln(M0/Mt) at this point (●). Conditions: [M] = 0.9 M, [M]/[CTA]/[VA-044] = 50/1/0.2. | ||
With encouraging results from the polymerisations at 65 °C, we set out to probe the “livingness” of the polymer before and after tdead and thus whether tdead was due to degradation of the RAFT agent. To this end, we isolated and purified two polymerisations of 1, one that had been stopped at intermediate conversions (ρ = 47%, t = 30 min), before tdead (Fig. S6A†) and one that was stopped at maximum conversion (ρ = 85%, t = 120 min), after tdead (Fig. S6B†). As expected, Boc-Px isolated before tdead was able to undergo complete chain extension with further addition of 1 and initiator (Fig. S6A†), thus demonstrating that at intermediate conversions the RAFT agent was still present in significant amounts. GPC analysis of Boc-Px isolated after tdead indicated that no chain extension had occurred, instead showing a bimodal distribution of molecular mass and high dispersities (Fig. S6B†) demonstrating that after tdead the RAFT group had been degraded. To probe if the RAFT agent degradation was temperature driven, we isolated and purified a second “living” Boc-Px at intermediate conversions (ρ = 52% t = 30 min) (Fig. S7†). This polymer was then heated for 90 minutes under standard polymerisation conditions, but this time without addition of 1 and initiator. We anticipated that heating the polymer this way should result in degradation of the RAFT agent, a hypothesis that was confirmed upon attempting to chain extend this terminated Boc-Px. In this case, high dispersities, together with a shoulder at high molecular mass, were observed, indicating that the Boc-Px which had been subjected to further heating was “dead” (Fig. S7†). Additional evidence of the RAFT agent degradation was obtained from NMR spectroscopy, where the protons associated with both the R and Z end group of the polymer chain could be observed for the “living” Boc-Px whereas “dead” Boc-Px showed a loss of the Z group (Fig. S8†).
Seeing how running the polymerisations at 65 °C gave the highest conversions (90%) at tdead of all the conditions evaluated, we decided to target different degrees of polymerisation using these conditions (Fig. 4). As before, targeting higher DPs resulted in slower rates of polymerisation, in particular for DP200 and DP300. While slower rates had a significant effect on the maximum conversion achieved (approx. 90%, 89%, 68% and 55% for DP 50, 100, 200 and 300 respectively), little effect was observed on the tdead, with most polymerisations “stopping” after 1 h (Fig. 4A).
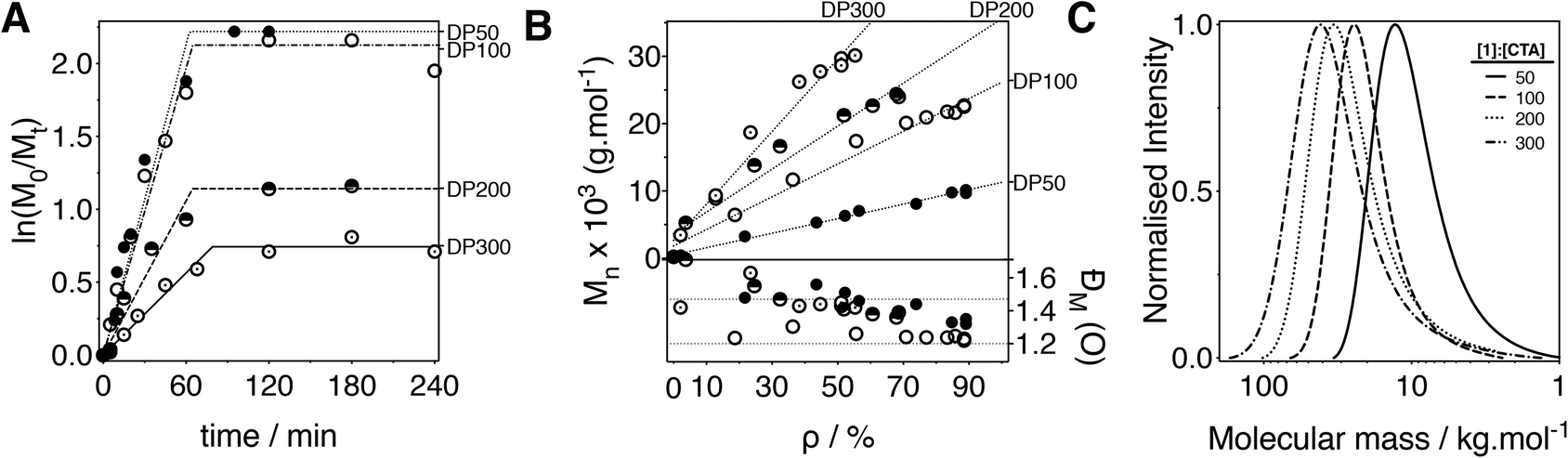 | ||
| Fig. 4 (A) Plot of fractional concentration of monomer ln(M0/Mt) vs. time. (B) Measured number average molecular mass (Mn) vs. conversion (ρ) (top) and dispersity in molecular mass (ĐM) vs. conversion (ρ) (bottom), for polymerisations of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1) performed at 65 °C with different 1:CTA ratios. (C) GPC chromatograms of the resulting polymers at the highest ρ obtained. Conditions: [M] = 0.9 M, [CTA]/[VA-044] = 5/1. Mn and ĐM calculated by GPC using 0.05 M LiBr in dimethylformamide (DMF) at 60 °C. | ||
Under these optimised conditions, the polymerisations retained features of a controlled polymerisation, with the molecular mass of the polymers increasing linearly with conversion, narrow dispersities in molar mass (Fig. 4C) and good end group fidelity if isolated before tdead. In all cases, the dispersities obtained were similar or lower to those reported previously.14 This improvement was particularly the case when targeting DPs of 100 and 200 with dispersities of <1.4 being observed at maximum conversion.
Conclusion
Here we have demonstrated the role of temperature and RAFT agent degradation in the polymerisation of N′-(tert-butoxycarbonyl)acryloyl hydrazide (1). Our results highlight that the polymerisation of this hydrazide monomer 1via RAFT can be severely hampered by the degradation of the chain transfer agent and that, under some circumstances, this degradation cannot be eliminated but rather outperformed if the rate of polymerisation is tuned. We demonstrate that by using a low-temperature initiator such as VA-044, optimal polymerisation conditions can be achieved at 65 °C. This way, poly(N′-(tert-butoxycarbonyl)acryloyl hydrazide)s with high degrees of polymerisation could be obtained while still maintaining low dispersities. We believe that further improvement of the polymerisation could be achieved through the choice of RAFT agents such as pyrazole or quaternised pyridinium dithiocarbamates,37,38 the use of photopolymerisation,39 or the use of alternative controlled polymerisation techniques. Our efforts in these directions will be reported in due course.Author contributions
PFT, OC and PA designed the work. OC, PA, GS and AR performed all the experimental work. OC, AR and PFT analysed the data. OC and PFT wrote the paper, with all other authors contributing to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest.Acknowledgements
All authors thank the P. F. T. group for useful discussions. P. F. T. thanks the University of Birmingham for the John Evans Fellowship. O. C. and P. A. thank BBSRC's Midlands Integrative Biosciences Training Partnership MIBTP (BB/M01116X/1) for PhD scholarships and A. R. thanks the European Union's Horizon 2020 Research and Innovation Programme (Marie Sklodowska-Curie Grant Agreement No. 795082).References
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- J. Li, F. Yu, Y. Chen and D. Oupický, J. Controlled Release, 2015, 219, 369–382 CrossRef CAS PubMed.
- J. J. Green and J. H. Elisseeff, Nature, 2016, 540, 386–394 CrossRef CAS PubMed.
- A. Muñoz-Bonilla and M. Fernández-García, Prog. Polym. Sci., 2012, 37, 281–339 CrossRef.
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 RSC.
- S.-J. Richards, M. W. Jones, M. Hunaban, D. M. Haddleton and M. I. Gibson, Angew. Chem., Int. Ed., 2012, 51, 7812–7816 CrossRef CAS PubMed.
- Z. Cao, L. Mi, J. Mendiola, J. R. Ella Menye, L. Zhang, H. Xue and S. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2602–2605 CrossRef CAS PubMed.
- Functional Polymers by Post-Polymerization Modification, ed. P. Théato and H.-A. Klok, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- J. Collins, Z. Xiao, M. Müllner and L. A. Connal, Polym. Chem., 2016, 7, 3812–3826 RSC.
- D. N. Crisan, O. Creese, R. Ball, J. L. Brioso, B. Martyn, J. Montenegro and F. Fernandez-Trillo, Polym. Chem., 2017, 1, 1392–1399 Search PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- E. R. L. Brisson, Z. Xiao, L. Levin, G. V. Franks and L. A. Connal, Polym. Chem., 2016, 7, 1945–1952 RSC.
- E. M. Muzammil, A. Khan and M. C. Stuparu, RSC Adv., 2017, 7, 55874–55884 RSC.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 9963–9965 CrossRef CAS PubMed.
- A. Kumar, R. R. Ujjwal, A. Mittal, A. Bansal and U. Ojha, ACS Appl. Mater. Interfaces, 2014, 6, 1855–1865 CrossRef CAS PubMed.
- J. M. Priegue, D. N. Crisan, J. Martínez-Costas, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Angew. Chem., Int. Ed., 2016, 55, 7492–7495 CrossRef CAS PubMed.
- A. Kumar, R. R. Ujjwal, A. Mittal, A. Bansal and U. Ojha, ACS Appl. Mater. Interfaces, 2014, 6, 1855–1865 CrossRef CAS PubMed.
- J. M. Priegue, I. Lostalé-Seijo, D. Crisan, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Biomacromolecules, 2018, 19, 2638–2649 CrossRef CAS PubMed.
- M. Juanes, O. Creese, P. Fernández-Trillo and J. Montenegro, MedChemComm, 2019, 526, 351–357 Search PubMed.
- J. D. Moskowitz, B. A. Abel, C. L. McCormick and J. S. Wiggins, J. Polym. Sci., Part A: Polym. Chem., 2015, 54, 553–562 CrossRef.
- B. A. Abel and C. L. McCormick, Macromolecules, 2016, 49, 465–474 CrossRef CAS.
- E. A. Hoff, B. A. Abel, C. A. Tretbar, C. L. McCormick and D. L. Patton, Polym. Chem., 2017, 8, 4978–4982 RSC.
- B. A. Chalmers, A. Alzahrani, G. Hawkins and F. Aldabbagh, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2123–2128 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 31, 4183 RSC.
- G. Moad, Prog. Polym. Sci., 2019, 88, 130–188 CrossRef CAS.
- Y. Zhou, Z. Zhang, A. Postma and G. Moad, Polym. Chem., 2019, 10, 3284–3287 RSC.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- Y. Zhou, J. He, C. Li, L. Hong and Y. Yang, Macromolecules, 2011, 44, 8446–8457 CrossRef CAS.
- T. M. Legge, A. T. Slark and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6980–6987 CrossRef CAS.
- J. Gardiner, I. Martinez-Botella, J. Tsanaktsidis and G. Moad, Polym. Chem., 2016, 7, 481–492 RSC.
- D. J. Keddie, C. Guerrero-Sanchez, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2011, 44, 6738–6745 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01222b |
| This journal is © The Royal Society of Chemistry 2019 |