
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First phosphorus AB2 monomer for flame-retardant hyperbranched polyphosphoesters: AB2vs. A2 + B3†
Jens C.
Markwart‡
 ab,
Alexander
Battig‡
c,
Thomas
Kuckhoff
a,
Bernhard
Schartel
*c and
Frederik R.
Wurm
*a
ab,
Alexander
Battig‡
c,
Thomas
Kuckhoff
a,
Bernhard
Schartel
*c and
Frederik R.
Wurm
*a
aPhysical Chemistry of Polymers, Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
bGraduate School Materials Science in Mainz, Staudinger Weg 9, 55128 Mainz, Germany
cBundesanstalt für Materialforschung und -prüfung (BAM), Unter den Eichen 87, 12205 Berlin, Germany
First published on 27th September 2019
Abstract
Branched polymers are an important class of polymers with a high number of terminal groups, lower viscosity compared to their linear analogs and higher miscibility, which makes them especially interesting for flame retardant applications, where the flame retardants (FR) are blended with another polymer matrix. Hyperbranched polyphosphoesters (hbPPEs) are gaining more and more interest in the field of flame retardancy, as low molar mass FRs often have the disadvantage of blooming out or leaching, which is not desired in consumer products. Here, we present the first phosphorus-based AB2 monomer for the synthesis of hbPPEs and assess its flame-retardant performance in an epoxy resin compared to a hbPPE synthesized by an A2 + B3 approach. The hbPPE synthesized from an AB2 monomer exhibited a slightly higher performance compared to a similar hbPPE, which was prepared by A2 + B3 polyaddition, probably due to its higher phosphorus content.
Introduction
Hyperbranched (hb) polymers, with their high number of terminal groups, lower viscosity, and higher matrix miscibility compared to their linear analogs, are especially interesting as flame-retardant additives, as effective blending with a polymer matrix is essential.1–5 Moreover, flame retardants (FRs) with complex architectures have a decreased impact on the material properties of polymers.6,7The synthesis of such dendritic polymers can be achieved by multi-step dendrimer syntheses, which are time-consuming, often need several purification steps and therefore unattractive for large scale, flame-retardant applications.1,8 In contrast, hb polymers are readily available by one polymerization step, e.g. by polycondensation of commercially available A2 + B3 monomer mixtures. hb polymers do not exhibit an architecture as perfect as dendrimers, because the polymers are statistically branched with structural and molar mass dispersities. Despite these architectural differences, hb polymers still retain many of the particular properties of dendrimers.1,3
Here, we present, to the best of our knowledge, the first phosphorus-based AB2 monomer for the synthesis of hb polyphosphoesters (hbPPEs), which are promising candidates as halogen-free flame-retardant additives.
To date, hbPPEs were synthesized by A2 + B3 approaches or by using AB* inimers, for which representative examples are given in Scheme 1. Penczek et al. prepared a family of oligomers with acidic end groups by an A2 + B3 approach by addition of H3PO4 to a bisphenol A based epoxy resins.9 Liu et al. described a water-soluble hbPPE through a self-condensing ring-opening polymerization of an AB* inimer (2-(2-hydroxyethoxy)ethoxy-2-oxo-1,3,2-dioxaphospholane).10 More recently, we used radical polyaddition of phosphorus-based B3-monomers with dithiols to prepare hbPPEs, polyphosphoramidates, and -amides to elucidate their decomposition mechanism as flame-retardant additives in epoxy resins.11
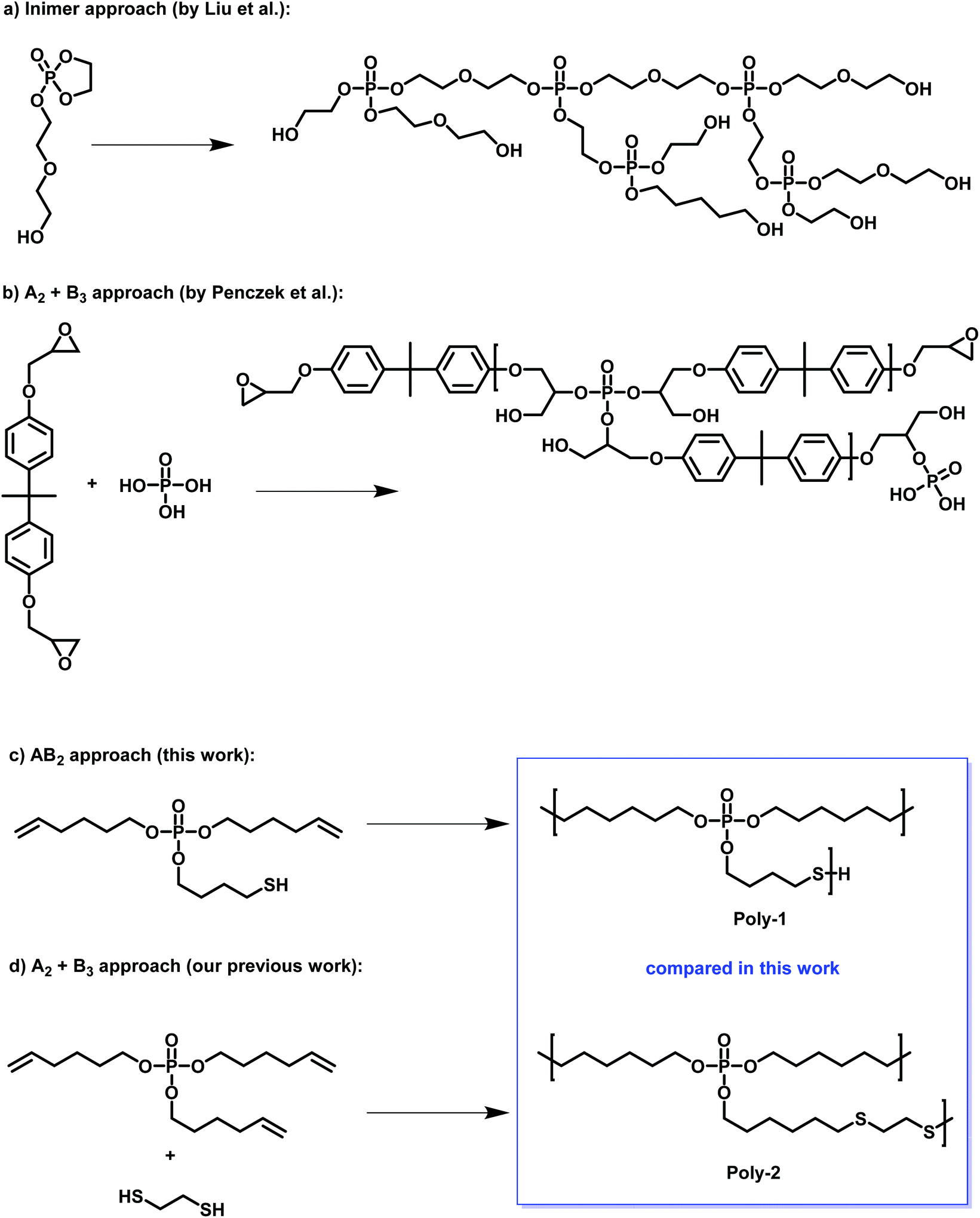 | ||
| Scheme 1 Examples of hbPPEs in literature: (a) hbPPEs via inimer approach by Yan et al.10 (b) A2 + B3 approach by Penczek et al.9 (c) this work: AB2 monomer and (d) the comparison A2 + B3 approach by Battig et al.11 | ||
Previous studies used hbPPEs due to their biocompatibility and biodegradability for mostly biomedical12,13 or optical applications.14 With the ban of some halogenated FRs, phosphorus-based derivatives as effective alternatives are in growing demand in recent years.15–18 Moreover, polymeric FRs are interesting as they exhibit less blooming out or leaching compared to low molar mass FRs, which is not desired in consumer products.19 In addition, the thermal stability of low molar mass FRs is usually lower, thus limiting their processability. In comparison, oligomeric or polymeric FRs exhibit increased thermal stability and therefore higher effectiveness, which leads to improved chemical interaction during decomposition, yielding higher char yields and better overall flame retardancy.11,20,21 Furthermore, FRs with different architectures have been investigated, stressing the impact of complex chemical structure on the mechanical properties and glass-transition temperature (Tg) of the polymer matrix.
With the first example of an AB2-type phosphate monomer for radical polycondensation, we present a straightforward approach to hbPPEs and thus avoid the chance of cross-linking during the synthesis. In addition, the versatile monomer design, which was exemplarily used for 1, allows further tuning of the P-content or the hydrophilicity, i.e. matrix compatibility, by variation of the alkyl-spacers, which makes the herein presented approach also applicable for other polymer matrices.
Results and discussion
For the synthesis of hyperbranched (hb) polymers, two common approaches exist: The ABn and An + Bm approach.22 In the ABn approach, first envisioned by Flory in 1952, only a single monomer with an ABn (n ≥ 2) structure is used.23 When A and B groups react selectively with each other, a statistically branched polymer without cross-linking is generated.24In the An + Bm approach, two monomers are used for polymerization (An and Bm (n, m ≥ 2)), with the most common method being the A2 + B3 approach, as several monomers are commercialized. However, to obtain soluble polymers, the polymerization needs to be terminated before the gel point, which requires adjustment of the reaction conditions, or adjustment of the monomer feed-ratio, etc. for each monomer set.24–26
For the synthesis of a hb polyphosphoester (hbPPE) via the ABn approach, an AB2 phosphoester with two different reactive groups (A and B) was prepared: di(hex-5-en-1-yl)(4-mercaptobutyl)phosphate (1) was synthesized in a four step reaction, starting from POCl3 and 4-bromobutan-1-ol (Scheme 2). 4-Bromobutan-1-ol was obtained from refluxing THF with HBr for several hours, followed by neutralizing with NaHCO3 and extraction with DCM, according to a literature protocol.27 An excess POCl3 was reacted with 4-bromobutan-1-ol. Removing the excessive amount of POCl3 gives compound 1a. 1a was used in the next step without further purification and treated with 5-hexen-1-ol to give compound 1b. The electrophilic alkyl bromide in 1b renders it a versatile precursor for various modifications like the introduction of other functional groups or as a monomer for ADMET itself. 1b was mixed with potassium thioacetate and stirred overnight to obtain compound 1c, which was purified by solvent extraction. The final AB2-monomer 1 for radical polyaddition was obtained after treating 1c with hydrazine, which cleaved the thioacetate group and released the free thiol. Monomer 1 is a liquid at room temperature and has a phosphorus content of 8.84 wt%. It is soluble in most organic solvents (e.g. toluene, tetrahydrofuran, ethyl acetate, acetone, dichloromethane and chloroform), but insoluble in water. It is important to mention that the butyl spacer between thiol and phosphorus is essential for the monomer stability: a similar monomer structure with an ethyl spacer was recently used to prepare linear PPEs with pendant 2-acetylthioethyl side chains.28 In this case, the pendant group acted as a protective group for the P-OH group after treatment with hydrazine, followed by a 3-exo-tet mechanism to release the P-OH group after cleavage of the phosphoester.
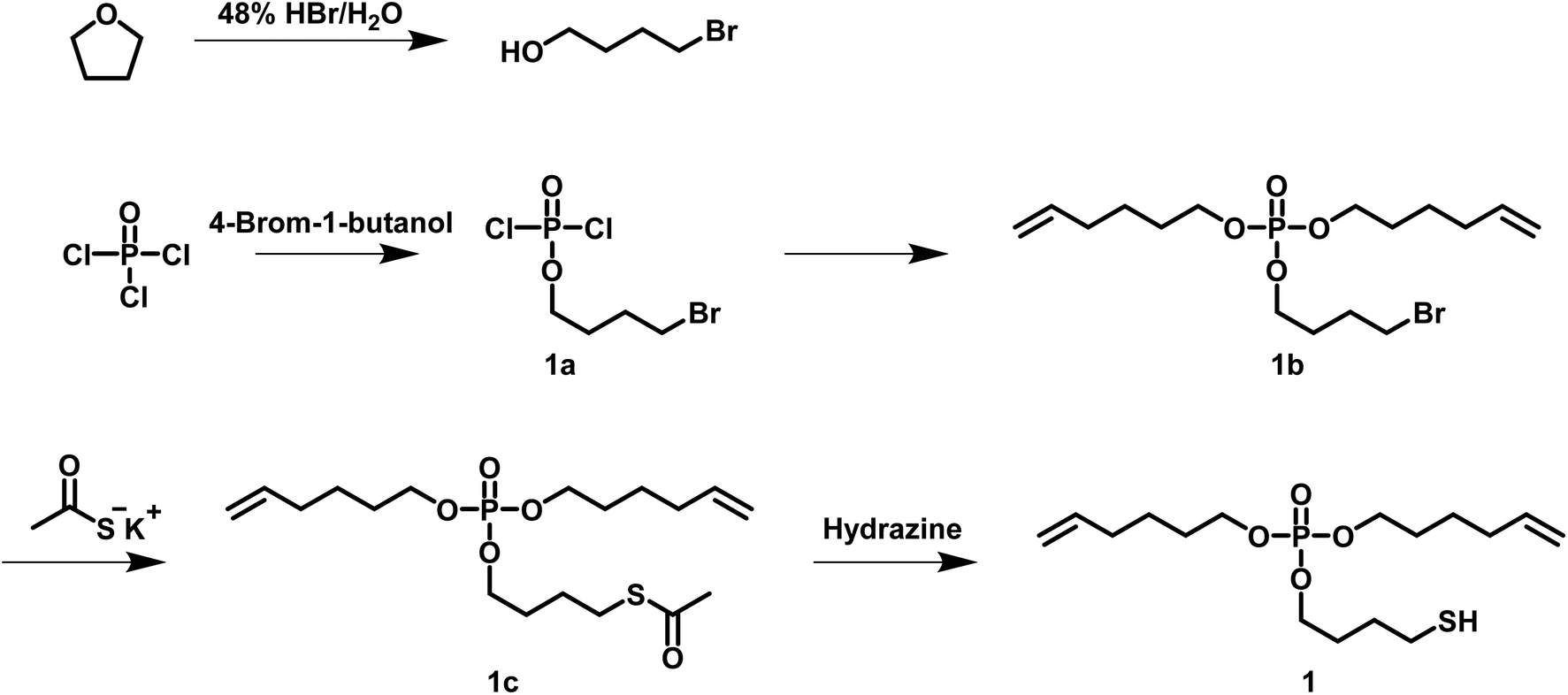 | ||
| Scheme 2 Synthesis scheme of di(hex-5-en-1-yl)(4-mercaptobutyl)phosphate (1). | ||
1H NMR spectroscopy (Fig. 1a) of 1 revealed two distinct resonances at 2.60 ppm (methylene group next to the thiol) and 1.36 ppm (SH). The olefinic signals were detected as multiplets in the region of 5.80 ppm and 5.00 ppm. The methylene group next to the double bond was found at 2.09 ppm and the methylene group next to the P–O group had a resonance at 4.05 ppm. The remaining signals of the methylene units were detected between 1.81 ppm and 1.45 ppm. The 31P NMR spectrum shows a single signal at −0.68 ppm (Fig. 1c).
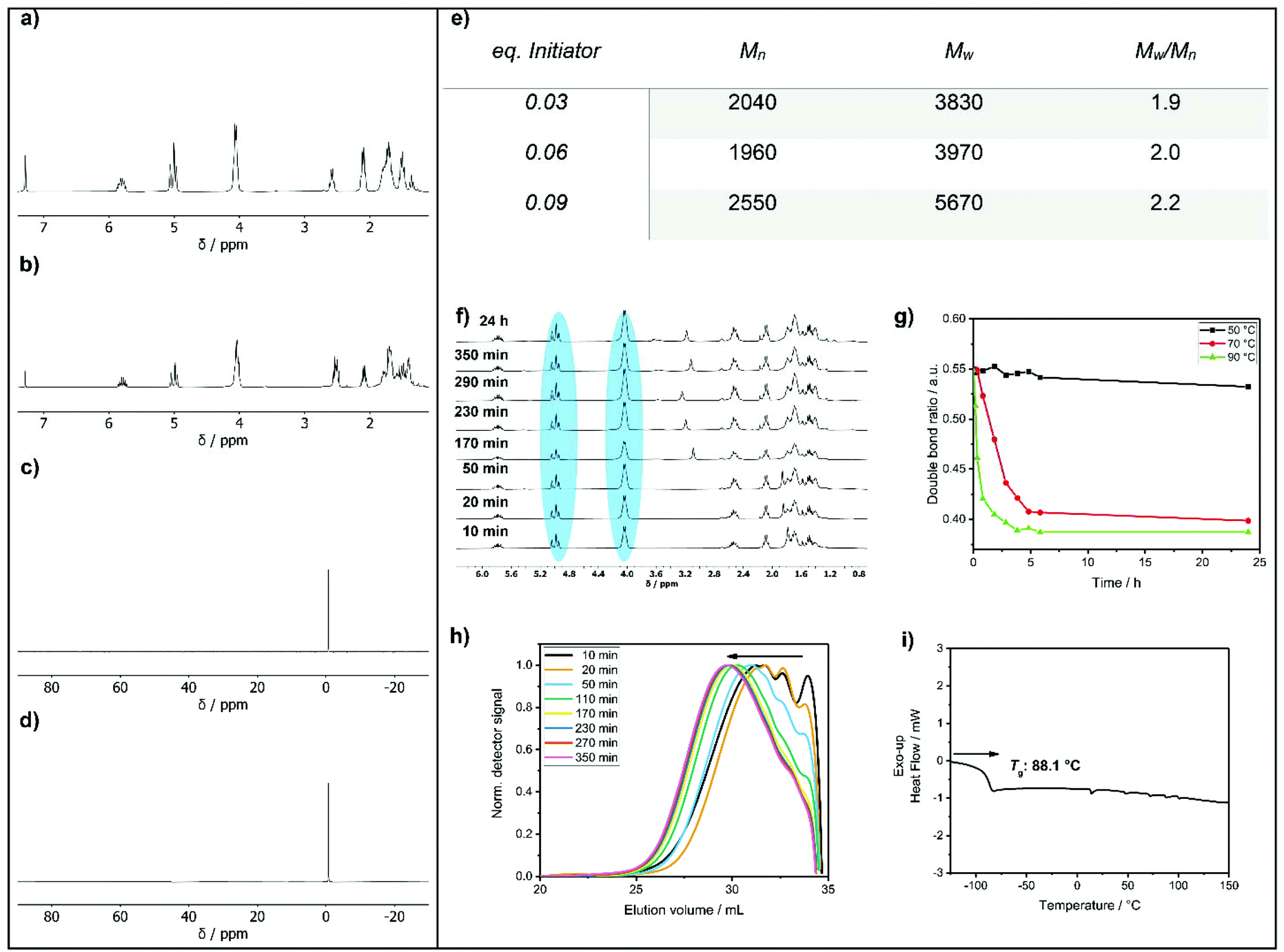 | ||
| Fig. 1 1H NMR spectra of 1 (a) and poly-1 (b); 31P NMR spectra of 1 (c) and poly-1 (d). (e) Molar masses of the polymer at different initiator equivalents; (f) 1H NMR kinetic of the polymerization of 1 at 90 °C and 0.03 eq. AIBN. (g) Polymerization kinetics measured by NMR (change of nDouble-bond/nEster over time) at different temperatures (50 °C, 70 °C and 90 °C). (h) GPC kinetics of the polymerization of 1 with 0.09 eq. AIBN (measured in DMF). (i) Differential scanning calorimetry (DSC) of poly-1 with a Tg at 88.1 °C. | ||
Compound 1 was used as AB2 monomer for the radical thiol–ene polyaddition to produce hbpoly-1 (Scheme 3). The statistically branched polymer with dendritic (D), linear (L), and terminal (T) units (cf.Scheme 3) was obtained as a viscous oil with a Tg of ca. −88 °C (Fig. 1i). The 1H NMR pattern of poly-1 was very similar to that of 1 (Fig. 1b); however, with increasing degree of polymerization, the olefinic resonances, the methylene group next to the S–H group and the S–H signal decreased. Due to signal overlap, calculation of a degree of branching was not possible. The polymerization was followed by GPC and NMR by taking samples throughout the reaction and calculating the ratio between the methylene groups next to the P–O (nEster) and the double bond (nDouble-bond) nDouble-bond/nEster (marked blue in Fig. 1f). During polymerization, a new resonance appeared at 2.54 ppm representing the methylene groups next to the thioethers. Polymerization was conducted at different temperatures (50 °C, 70 °C, and 90 °C) with 0.03 eq. AIBN and additionally with different amounts of AIBN (0.03 eq., 0.06 eq. and 0.09 eq.) at 70 °C (e.g.Fig. 1h). From the NMR data, only very slow reaction kinetics were detected at 50 °C, while at elevated temperatures (70 °C), no further reaction was observed after 7 h (Fig. 1g). With increasing initiator concentration, a slight increase in molar mass was observed (Fig. 1e). The 31P NMR resonance of poly-1 remained relatively unchanged compared to the monomer with a single signal at −0.70 ppm (Fig. 1d). As the degree of polymerization increased, nDouble-bond/nEster became smaller.
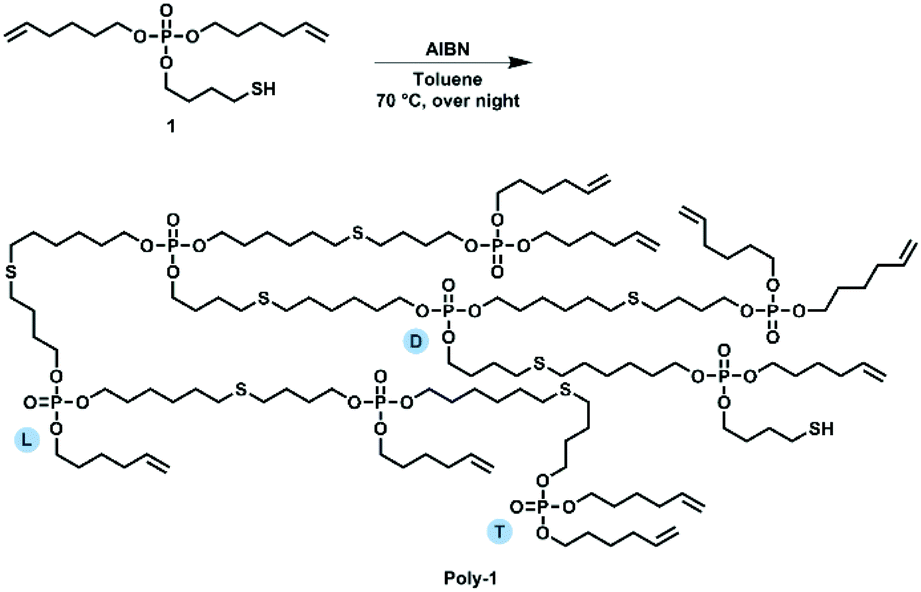 | ||
| Scheme 3 Hyperbranching polymerization of monomer 1 to poly-1 by radical thiol–ene polyaddition. | ||
For the flame retardancy investigations, the resulting polymer had an Mw of 5500 g mol−1 with an Mw/Mn of 2.39 (GPC in DMF). Poly-1 was used as an additive flame retardant (FR) in epoxy resins, and the FR properties were compared to a similar hbPPE prepared by the A2 + B3 approach (poly-2, Scheme 1), Mw of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1 with an Mw/Mn of 3.29 (GPC in THF),11 and a commercial phosphate-based FR, namely bisphenol A bis(diphenyl phosphate) (BDP), which was already used in epoxy resins like DGEBA/DMC.19,29 The ratio nDouble-bond/nEster, an indication of the amount of terminal double bonds, was identical for poly-1 and poly-2 (0.39). Looking at the phosphorus content, poly-1 exhibits the same amount of P when compared to the monomer. In contrast, for poly-2 the P content varied, depending on the monomer ratio and workup procedure. The theoretical phosphorus content deviated from the measured phosphorus content after precipitation. The theoretical phosphorus content of poly-2 is 5.1 wt%, the measured phosphorus content by elemental analysis is 7.7 wt%.
300 g mol−1 with an Mw/Mn of 3.29 (GPC in THF),11 and a commercial phosphate-based FR, namely bisphenol A bis(diphenyl phosphate) (BDP), which was already used in epoxy resins like DGEBA/DMC.19,29 The ratio nDouble-bond/nEster, an indication of the amount of terminal double bonds, was identical for poly-1 and poly-2 (0.39). Looking at the phosphorus content, poly-1 exhibits the same amount of P when compared to the monomer. In contrast, for poly-2 the P content varied, depending on the monomer ratio and workup procedure. The theoretical phosphorus content deviated from the measured phosphorus content after precipitation. The theoretical phosphorus content of poly-2 is 5.1 wt%, the measured phosphorus content by elemental analysis is 7.7 wt%.
Pyrolysis: thermal decomposition of FRs via TGA
The pyrolytic decomposition of the FRs was investigated using thermogravimetric analysis (TGA) (Fig. 2). During burning, the thermal decomposition of the material feeds volatile fuel into the flame zone, where exothermal combustion reactions, i.e. oxidation, occur. However, at the solid/gas interface, the reactions in the anaerobic pyrolysis zone determine the fire behavior. This model is accurate for polymeric materials in developing fires, which are simulated in the cone calorimeter, but also reaction-to-small-flame tests such as UL94 and LOI. Therefore, investigations into the pyrolytic reactions of FRs and FR-containing polymers via TGA in nitrogen atmosphere are an important analytical tool to understand the chemical mechanisms underlying the FR's modes of action.30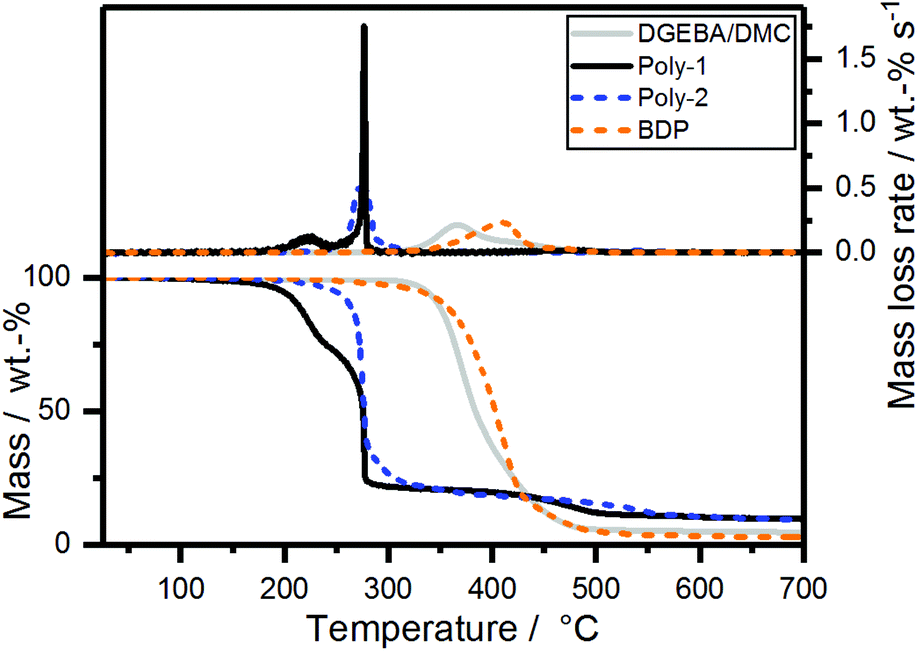 | ||
| Fig. 2 Mass loss (bottom) and mass loss rate (top) over T of poly-1, poly-2, bisphenol A bis(diphenyl phosphate) and neat epoxy resin from TGA measurements (10 K min−1; N2). | ||
The mass loss curve of poly-2 exhibited a main single decomposition step at 274 °C with a gradual decomposition thereafter (Fig. 2). Poly-1 exhibited an additional decomposition step at ca. 226 °C, followed by the second decomposition step at the same temperature as poly-2. The additional decomposition step might be rationalized with the cleavage of the terminal alkyl-SH group, similar to the mechanism described previously by Markwart et al.,28 which was confirmed by the presence of tetrahydrothiophene (from pyrolysis-(Py)-GC/MS measurements at 250 °C (Fig. S6 and S8†) and TGA-FTIR measurements (Fig. S16†)). The amount of residue at 700 °C was very similar for both polymers (poly-1: 9.7 wt%, poly-2: 9.3 wt%).
Pyrolysis: evolved gas analysis of FRs via TG-FTIR
Evolved gas analysis during pyrolysis of poly-1 was performed via Py-GC/MS and TG-FTIR measurements. The analysis of epoxy resin (EP) and poly-2 has been previously described in detail and will therefore not be discussed herein.11,31 For poly-1, two single-shot Py-GC/MS measurements at varied pyrolysis temperatures (250 °C and 500 °C) were conducted to isolate the decomposition products in the first decomposition step. The gas chromatogram at 250 °C (Fig. S6†) displays a single large signal at 5.62 min retention time, while at 500 °C (Fig. S7†), additional signals between 2.60–3.56 min were detected, as well as minor signals >5.62 min. The mass spectrum at 5.62 min was identified as tetrahydro thiophene (Fig. S8 and S9†), thus confirming the cleavage of alkyl-SH groups of poly-1 during the first decomposition step seen in TGA. The signals between 2.60–3.56 min corresponded to 1,5-hexadiene (Fig. S10 and S11†) and its thermal rearrangement products. The rearrangement is proven by the presence of cyclohexane (Fig. S12 and S13†) at 3.56 min, a product of cyclization of 1,5-hexadiene. At 6.57 min, the signal was identified as 5-hexen-1-ol (Fig. S14 and S15†) resulting from hydrolysis of the phosphate-moiety.Pyrolysis: EP-FR preparation and material properties
The FR-performances of BDP, poly-1, and poly-2 were studied in an epoxy resin (EP) based on bisphenol A diglycidylether (DGEBA) and 2,2′-dimethyl-4,4′-methylene-bis-(cyclohexylamine) (DMC). All epoxy plates were prepared in the following manner: DGEBA was mixed with the respective FR (loading: 10 wt%) until homogenous. Then, DMC was added, and the mixture was poured into appropriately sized aluminum molds, followed by curing for 30 min at 90 °C, 30 min at 120 °C, and 1 h at 150 °C. Additive FRs can act as plasticizers in epoxy resins, thus reducing the glass transition temperature (Tg) of the resulting composite. Differential scanning calorimetry (DSC) measurements revealed that the flame retardant containing epoxy resins (EP-FRs) lowered Tg by an average of 24 °C: Poly-1 had the strongest impact on the Tg of EP, lowering it by about 30 °C to 124 °C (Tg, EP = 155 °C), while poly-2 and BDP had a similar impact on the Tg of EP (Tg, EP-poly2 = 132 °C; Tg, EP-BDP = 133 °C). The impact of FRs on the Tg of EP is presented in Fig. S20,† and the change of Tg relative to EP is noted.Pyrolysis: thermal decomposition and evolved gas analysis of EP-FRs via TGA and TG-FTIR
The decomposition behavior of EP-FRs was investigated by analyzing the mass loss and evolved gas during pyrolytic decomposition via TGA coupled with FTIR (Table S2†). A significant change in decomposition behavior was observable for all EP-FRs, as the mass loss and mass loss rate curves (Fig. 3) and the change in residue yields at 700 °C proved. The pure epoxy decomposed with an onset temperature (T5%) of 338 °C and reached the temperature of maximum mass loss rate (Tmax) at 372 °C. EP decomposed in a single main step with a mass loss equal to 62 wt%. Following the main decomposition step, a shoulder beginning at 424 °C with a mass loss of 33 wt% was observed. At 700 °C, the residue yield was 4.5 wt%. The mass loss and evolved gas analysis of the epoxy resin (DGEBA/DMC) has been extensively investigated; therefore, it will not be discussed further.32,33 When BDP was added to the resin (EP-BDP), the composite decomposed similarly to the pure EP, but T5% was lowered by about 33 °C, and Tmax was 15 °C lower than EP. This change is attributed to a reduction in cross-linking density of the EP-system when additives are present.34 The plateau which started at 423 °C exhibited a lower decomposition rate compared to pure EP. An increase in mass loss at Tmax to 75 wt% and a decrease to 16 wt% at the shoulder was observable. An explanation for this phenomenon is the interaction of the FR with the decomposing matrix.35 More specifically, the phenol-derivates and cycloalkanes are bound; these exhibit a production rate maximum in this temperature range.36 As a result, the residue yield of EP-BDP increased to 8.1 wt%, which is nearly twice that of pure EP.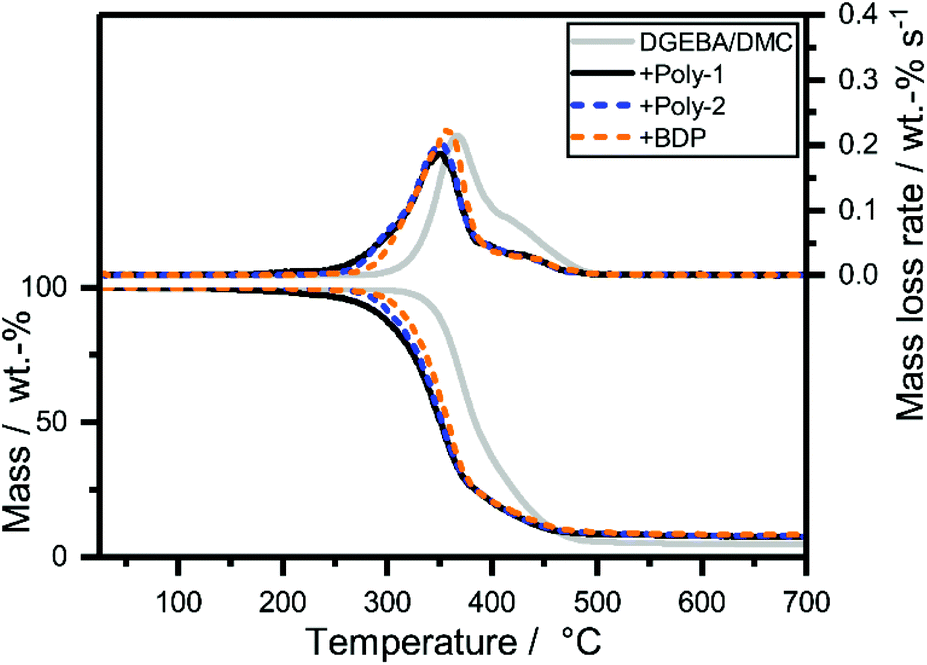 | ||
| Fig. 3 Mass loss (bottom) and mass loss rate (top) over T of EP-FRs from TGA measurements (10 K min−1; N2). | ||
All hb-FR containing EPs (EP-hb-FRs) exhibited a decomposition behavior similar to EP-BDP. The T5% of EP was lowered by 70 °C for poly-1 and 47 °C for poly-2. Tmax was also lowered when FRs were present, on average by approx. 16 °C. The lower T5% of EP-poly-1 compared to EP-poly-2 is caused by the additional decomposition step in poly-1. All residue yields of EP-FRs were in a similar range, and all investigated FRs increased the residue of EP (7.9 and 7.7 wt% for poly-1 and poly-2, respectively, and 8.1 wt% for BDP). The neat EP had a residue yield of only 4.6 wt%. This increase in residue indicates that the tested FRs interact with the decomposing matrix. As a result, thermally stable residues are formed.
From the evolved gas analysis of EP-FRsvia TG-FTIR (Fig. S17†), two distinct decomposition products were visible for EP-poly-1 and EP-poly-2, the first appearing in the range of about 290 °C and the second between 360–380 °C. At ca. 290 °C, the spectra of EP-poly-1 and EP-poly-2 exhibited the evolution of 5-hexen-1-ol, a product of either hydrolysis or transesterification. Its presence indicates that hb-FRs are active near T5%, forming either lower molecular phosphates (hydrolysis) which are active in the condensed phase, or phosphorylating the polymer matrix (transesterification), thus forming char precursors. The condensed phase mode of action is additionally proven by hot-stage FTIR (Fig. S19†). At 360–380 °C, all spectra are identical to EP, as the matrix decomposes in this temperature range.
Pyrolysis: condensed phase analysis of EP-FRs via hot-stage FTIR
The condensed phase mode of action of poly-1 and poly-2 in EP was proven by hot-stage FTIR measurements (Fig. S18 and S19†). Fig. S18† displays the unique signals of EP-poly-1 at 300 °C, namely 1146 and 1108 cm−1, which are shifted to slightly lower wavenumbers and increase in intensity at 500 °C (Fig. S19†). These signals may correspond to ν(P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of R2–(PO)–OH resulting from the cleavage of terminal alkyl-SH groups which occurs more readily than cleavage of terminal hexene-moieties, as observed in Py-GC/MS (Fig. S8†) and TG-FTIR measurements (Fig. S16†) of poly-1. It is conceivable that poly-1 is more reactive than poly-2 in terms of phosphorylation of the epoxy resin matrix, especially given the increase in residue yields of fire testing and pyrolysis (Tables S1 and S2,† respectively). At 600 °C, the hot-stage FTIR spectrum of EP-poly-1 exhibits many bands that are also present in EP-poly-2 and EP-BDP, which have already been previously described as phosphorus signals.11 Moreover, additional bands at 1400, 1125, 1010, 974, and 585 cm−1 are present. Many types of compounds, including vinylene-moieties, cyclic aliphatic hydrocarbons, and secondary or tertiary alcohols, present signals at these wavenumbers. While the identification of specific compounds in hot-stage FTIR is not always possible, it is certain that the spectrum of EP-poly-1 presents clear signals that are distinct and different from EP, thereby proving a condensed-phase spectrum of poly-1 in EP.
O) of R2–(PO)–OH resulting from the cleavage of terminal alkyl-SH groups which occurs more readily than cleavage of terminal hexene-moieties, as observed in Py-GC/MS (Fig. S8†) and TG-FTIR measurements (Fig. S16†) of poly-1. It is conceivable that poly-1 is more reactive than poly-2 in terms of phosphorylation of the epoxy resin matrix, especially given the increase in residue yields of fire testing and pyrolysis (Tables S1 and S2,† respectively). At 600 °C, the hot-stage FTIR spectrum of EP-poly-1 exhibits many bands that are also present in EP-poly-2 and EP-BDP, which have already been previously described as phosphorus signals.11 Moreover, additional bands at 1400, 1125, 1010, 974, and 585 cm−1 are present. Many types of compounds, including vinylene-moieties, cyclic aliphatic hydrocarbons, and secondary or tertiary alcohols, present signals at these wavenumbers. While the identification of specific compounds in hot-stage FTIR is not always possible, it is certain that the spectrum of EP-poly-1 presents clear signals that are distinct and different from EP, thereby proving a condensed-phase spectrum of poly-1 in EP.
Fire testing: forced flaming conditions
All EP-hb-FRs reduced the fire load (THE) of EP. Poly-1 decreased the fire load of EP by 21%, whereas poly-2 decreased the fire load by 17% and BDP by 19%. Poly-1 reduced THE of EP more strongly than poly-2 because it was able to retain more fuel in the condensed phase, as evidenced by its higher residue yield (Fig. 4d). This fuel retention may be caused by the higher reactivity of poly-1 compared to poly-2: as poly-1 has a lower T5%, its decomposition products may interact earlier with the decomposing matrix than poly-2, therefore increasing char yield, i.e. fuel fixation, and thus lowering THE. The tested FRs lowered the peak of heat release rate (PHRR) of EP by ca. 30% for BDP and poly-1, and by 44% for poly-2. The HRR curves (Fig. 4a) indicate that the formation of a protective char layer on the sample surface resulted in a plateau-like shape approx. 30 s after ignition, reducing PHRR by shielding the underlying material from irradiation. All FRs increased residue yields in the order EP-poly-1 > EP-poly-2 > EP-BDP. EP-Poly-1 exhibited the highest residue amount (11.5 wt%) and EP-BDP showed the lowest (3.1 wt%). The residues after fire testing (Fig. 4d) help visualize the differences in fire performance of poly-1 and poly-2 in EP: The residue of EP-poly-2 was more voluminous than that of EP-poly-1, pointing to higher gas emission, similar to intumescent FR systems. This large char volume was effective in shielding some of the underlying material, acting as a protective layer and thereby reducing PHRR of EP.37 However, EP-poly-1 exhibited a lower THE and higher residue yield than EP-poly-2, because the thermal properties of its char were greater. As a result, poly-1 was better able to bind fuel in the condensed phase in the form of char, which increased residue yields and thus reduced the fire load, i.e. THE. Although EHC of both materials were nearly identical, the change in char characteristics explains the difference in fire performance, as the residue morphology and its properties often determine effective flame retardancy.35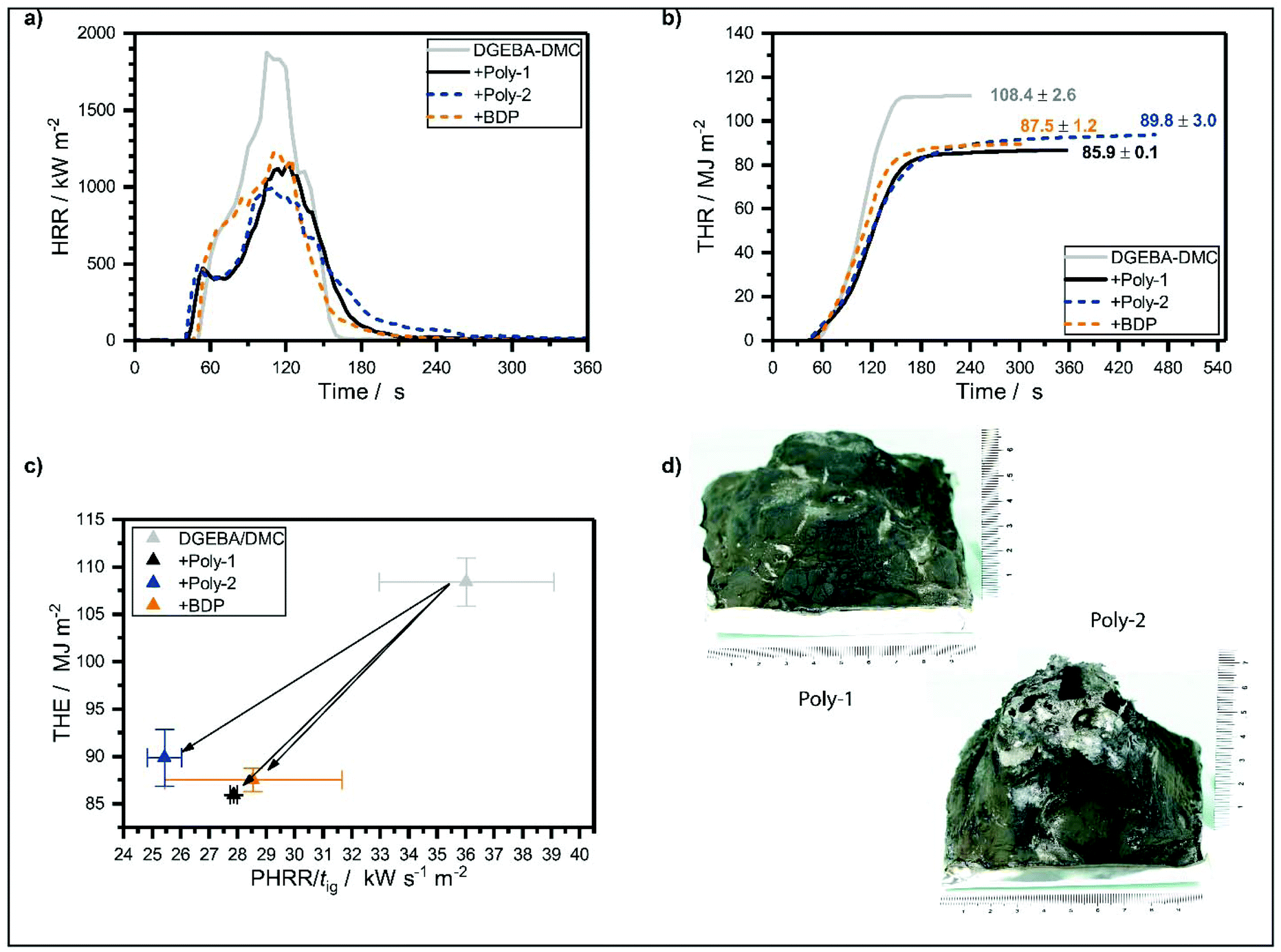 | ||
| Fig. 4 (a) Heat release rate (HRR) of epoxy resin and epoxy resin with FRs. (b) Total heat released (THR) of epoxy resin and epoxy resin with FRs. (c) Petrella plot of the different epoxy resins with all FRs having a positive effect (lowering THE and PHRR/tig). (d) Char residues of EP-poly-1 and EP-poly-2 after cone calorimeter test. Residue of EP-poly-2 is more voluminous than that of EP-poly-1, pointing to higher gas emission, similar to intumescent FR systems. | ||
Additionally, the effective heat of combustion (EHC) was reduced by the release of P-containing volatiles. These P-containing volatiles acted in the gas phase as radical scavengers, i.e. by lowering the concentration of highly reactive radicals (H˙, HO˙, CO˙, etc.) through P˙ or PO˙ radicals. The resulting flame inhibition led to less complete combustion; additionally, P enhanced charring thus stored fuel in the condensed phase, all of which resulted in a reduction of THE (Fig. 4b).
To further assess fire behavior and flame retardancy, the fire load (THE) is often plotted against the fire growth index (PHRR/tig, Fig. 4c),38 because THE describes heat release quantitatively, however it does not describe the release rate. PHRR/tig is a means of describing the time-dependent flashover potential or fire growth index, i.e. the severity of a fire, or peak heat release potential; however, it is not quantitative. The investigated FRs reduced both PHRR/tig and THE of EP, which had a PHRR/tig of 36 kW m−2 s−1 and a THE of 110 MJ m−2. EP-Poly-1 had a performance similar to EP-BDP: BDP reduced THE of EP by 19% to 88 MJ m−2 and poly-1 reduced it by 21% to 86 MJ m−2. The PHRR/tig was reduced by 21% to 29 kW s−1 m−2 and by 23% to 28 kW s−1 m−2 for BDP and poly-1, respectively. Poly-2 exhibits a higher THE compared to poly-1 and BDP but has a stronger reduction in PHRR/tig. The graph visualizes the overall good flame-retardancy potential of the hb-FRs: a shift to the lower-left corner of the coordinate system indicates a reduction of overall heat and fire growth. Both hb-FRs lower both values on a similar level to the benchmark material, proving their efficacy for this polymer resin system. Moreover, poly-1 exhibited lower fire loads than poly-2 in EP; this implies that poly-1 was more able to bind fuel in the condensed phase, as proven by the higher overall char yield. On the other hand, poly-2 reduced the fire growth rate more strongly than poly-1 in EP: this resulted from the better thermal barrier properties of EP-poly-2's char, which lowered PHRR, as well as the higher thermal stability of poly-2 which led to an increased tig.
Conclusion
Hyperbranched polymers, especially polyphosphoesters, are interesting candidates as polymeric flame retardants. hbPPEs were previously prepared by An + Bm approaches, which might result in cross-linked PPEs, as adjustment of the monomer feed-ratio or termination before the gel point needs to be carefully conducted. Herein, we presented the first phosphorus-based AB2 monomer (1), allowing the synthesis of hbPPEs in a single polyaddition step without the chance of undesired cross-linking.In addition to the simplified polymerization procedure, poly-1 exhibited a slightly higher performance compared to similar hbPPEs (poly-2), prepared by A2 + B3 polyaddition, probably due to its higher phosphorus content. This work further extends the possibilities for the preparation of branched polyphosphoesters, which might be used in biofriendly flame retardant applications or biomedical applications.
Experimental section
Materials
All chemicals were purchased from commercial suppliers as reagent grade and used without further purification.Samples for TGA-FTIR and hot-stage FTIR were milled prior to use. Powdered specimens were obtained using a RETSCH CryoMill under liquid nitrogen cooling.
DSC
For Differential Scanning Calorimetry (DSC), a Mettler Toledo DSC 823e was used. With a heating and cooling rate of 10 K min−1, three measurements of heating, cooling and heating were performed. The measurements were done in a nitrogen atmosphere with a flow rate of 30 mL min−1.EP-FRs were measured on a Netzsch 204 F1, type Pheonix. Two cooling and three heating runs were performed on 5 mg bulk material samples; the rate was 10 K min−1, the temperature range was −80 to 180 °C, and the nitrogen flow rate was 30 mL min−1. The second and third heating rate were used to determine Tg.
TGA
For the thermogravimetric analysis (TGA) of the neat flame retardants, a Mettler Toledo TGA/DSC 3+ in a nitrogen atmosphere was used. Using 10 mg of the sample, the measurements were performed in a range from 25 °C to 700 °C with a heating rate of 10 K min−1.TG-FTIR
Pyrolysis investigations into mass loss and evolved gas analysis were performed via thermogravimetric analysis (TGA) on a Netzsch TG 209, type Iris, which was coupled via transfer line to a Bruker Tensor 27 infrared spectrometer (FTIR). For TG-FTIR measurements of EP and EP-FRs, 10 mg powdered samples were used, while for pure hb-FRs, 5 mg samples were measured. Measurements were conducted from 30–900 °C (10 K min−1) under a 30 ml min−1 nitrogen purge. Evolved gases passed through a transfer line heated to 270 °C into the FTIR gas cell which was also heated to 270 °C. The measuring range was 4000–400 cm−1 with a resolution of 1 cm−1.Hot stage FTIR
Pyrolysis investigations of the condensed phase activity were performed on a Bruker Vertex 70 FTIR equipped with a Linkam FTIR600 hot stage cell. Powdered EP and EP-FR samples (ca. 5 mg) were mixed with 150 mg KBr in a mortar and pestle, then pressed into a platelet at 7 bar. Specimens were heated from 30–600 °C at a rate of 10 K min−1 under a nitrogen atmosphere. The measuring range was 4000–400 cm−1 with a resolution of 0.4 cm−1.Pyrolysis-gas chromatography-mass spectrometry (Py-GC/MS)
Pyrolytic evolved gas analysis was performed on a pyrolysis-gas chromatograph/mass spectrometer using a Frontier Lab PY3030iD micro-furnace single-shot pyrolyzer connected to an Agilent Technologies 7890B gas chromatograph via a split-/splitless inlet port. An Agilent Technologies 7890B mass selective detector was combined with the gas chromatograph; the ionization energy (EI) was 70 eV and the scan range was 15–550 amu. 150 μg samples were pyrolyzed under helium atmosphere and inserted into the pyrolyzer via gravimetric fall; the temperature was 500 °C, except for measurements of poly-1, where the pyrolyzer temperature was set to 250 °C for an additional measurement. All evolved pyrolysis products were separated under a helium flow of 1 mL m−1 in an Ultra Allow+- 5 capillary column with a length of 30 m, inner diameter of 0.25 mm, and film thickness of 0.25 μm. First, the column was heated to 40 °C and held there for 2 min, then heated at a rate of 10 K min−1 up to 300 °C, where it was kept for 10 min. The GC injector was operated in a split mode of 1:300; the interface temperature was 300 °C. MS peak assignments were made using the NIST 14 MS library.
Cone calorimeter
Fire testing was conducted on an FTT cone calorimeter operating at a heat flux of 50 kW m−2 according to ISO 5660, simulating a developing fire.39 Samples sized 100 mm × 100 mm × 4 mm were conditioned at 23 °C and 50% RH for at least 48 h, then measured at a distance of 35 mm from the cone heater, as a distance of 25 mm was not suitable for the large residues of the materials.40GPC
GPC measurements were performed in DMF (+LiBr 1 g L−1) with a PSS SecCurity system (Agilent Technologies 1260 Infinity). Sample injection was performed by a 1260-ALS autosampler (Waters) at 60 °C. SDV columns (PSS) with dimensions of 300 × 80 mm, 10 μm particle size, and pore sizes of 10000, 1000, and 100 Å were employed. The IR 1260 RID detector and UV-vis 1260-VWD detector (Agilent) were used for detection. Calibration was achieved using poly(styrene) standards provided by Polymer Standards Service.
Elemental analysis
Elemental analysis was run on an Elementar Vario EL cube.NMR
Nuclear magnetic resonance (NMR) analysis, 1H, 31P {H} and 13C {H} NMR spectra were recorded with Bruker Avance spectrometers operating with 250, 300, 500 and 700 MHz frequencies in deuterated chloroform, deuterated dimethyl sulfoxide or deuterated N,N-dimethylformamide as a solvent. The calibration of the spectra was done against the solvent signal. The spectra were analyzed using MestReNova 9 from Mestrelab Research S.L.4-Bromobutan-1-ol
The synthesis was done according to a literature procedure.27 In a 1 L flask, THF (270 mL, 3.33 mol) was added to hydrobromic acid (48%, 180 g, 1.06 mol). The mixture was refluxed for two hours, transferred into an Erlenmeyer flask, and the reaction was neutralized by the addition of NaHCO3 under strong CO2 development. The aqueous solution was extracted with dichloromethane and the organic layers were combined and dried with Na2SO4. The solvent was removed at reduced pressure, yielding the product (47.3 g, 29%), which was used without further purification.1H NMR (250 MHz, CDCl3): δ [ppm] = 4.01 (s, 1H), 3.69 (t, J = 6.4 Hz, 2H), 3.44 (t, J = 6.6 Hz, 2H), 2.05–1.79 (dd, J = 8.0, 6.6 Hz, 2H), 1.79–1.62 (dd, J = 8.5, 6.4 Hz, 2H).
4-Bromobutyl phosphorodichloridate (1a)
To a dried three-necked, 500 mL round bottom flask equipped with two 100 mL dropping funnels, 0.522 mol phosphoryl chloride (80.00 g, 47.62 mL, 521.78 mmol, 5.0 eq.) were added to ice-cooled, dry toluene (100 mL) under argon atmosphere. 4-bromobutan-1-ol (15.97 g, 9.51 mL, 104.36 mmol, 1.0 eq.) dissolved in dry toluene (50 mL) and pyridine (8.25 g, 8.42 mL, 104.36 mmol, 1.0 eq.) dissolved in dry toluene (50 mL) were added to the above flask dropwise, keeping the temperature at 0 °C. After stirring overnight at room temperature, pyridine hydrochloride was removed as a white solid by filtration. The filtrate containing the alkylene dichlorophosphate in toluene was concentrated at reduced pressure. 4-Bromobutyl phosphoro-dichloridate was obtained as a colourless liquid (yield: 21.6 g, 77%).1H NMR (300 MHz, 298 K, CDCl3, δ/ppm): 4.43–3.34 (m, 2H), 3.49–3.43 (t, J = 5.8 Hz, 2H), 2.07–1.95 (m, 4H).
31P{H} NMR (202 MHz, 298 K, CDCl3, δ/ppm): 7.21.
4-Bromobutyldi(hex-5-en-1-yl) phosphate (1b)
To a dry three-necked, 500 mL round bottom flask fitted with a 250 mL dropping funnel, 5-hexen-1-ol (17.61 g, 21.11 mL, 175.29 mmol, 2.1 eq.) and pyridine (13.87 g, 14.15 mL, 175.29 mmol, 2.1 eq.) were added to dry toluene (100 mL) under an argon atmosphere. 1 (22.53 g, 83.47 mmol, 1.0 eq.) dissolved in dry toluene (100 mL) was added to the above flask dropwise at room temperature. After stirring overnight, pyridine hydrochloride was removed as a white solid by filtration. The organic solution was washed with sodium bicarbonate solution, 10% hydrochloric acid, and sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was removed at reduced pressure. 4-Bromobutyldi(hex-5-en-1-yl) phosphate was obtained as a yellow liquid (yield: 30.2 g, 88%).1H NMR (300 MHz, 298 K, CDCl3, δ/ppm): 5.90–5.68 (m, 2H), 5.06–4.92 (m, 4H), 4.40–3.92 (m, 6H), 3.69–3.61 (t, J = 6.3 Hz, 2H), 3.48–3.40 (t, J = 6.3 Hz,2H), 2.21–2.05 (m, 4H), 2.05–1.74 (m, 4H), 1.74–1.59 (m, 4H), 1.53–1.41 (m, 4H). 31P{H} NMR (202 MHz, 298 K, CDCl3, δ/ppm): −0.71.
Di(hex-5-en-1-yl)(4-acetylthiobutyl)phosphate (1c)
To a dry one-necked, 500 mL round bottom flask, 2 (29.05 g, 73.11 mmol, 1.0 eq.) and potassium thioacetate (9.18 g, 80.42 mmol, 1.1 eq.) were dissolved in acetone (100 mL). After stirring overnight at room temperature, potassium bromide was removed as a white solid by filtration. Acetone was removed at reduced pressure and the residue was dissolved in toluene (100 mL). The mixture was washed with sodium bicarbonate solution, 10% hydrochloric acid and sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate, filtered and the solvent was removed at reduced pressure to isolate S-(4-((bis(hex-5-en-1-yloxy)phosphoryl)oxy)butyl)ethanethioate (yield: 27.7 g, 96%).1H NMR (300 MHz, 298 K, CDCl3, δ/ppm): 5.87–5.70 (m, 2H), 5.06–4.92 (m, 4H), 4.23–3.94(m, 6H), 3.92–2.82 (m, 4H), 2.32 (s, 3H), 2.13–2.02 (m, 4H) 1.79–1.63 (m, 8H), 1.54–1.42 (m 4H).
31P{H} NMR (202 MHz, 298 K, CDCl3, δ/ppm): −0.55.
Di(hex-5-en-1-yl)(4-mercaptobutyl)phosphate (1)
1c (26.78 g, 68.24 mmol, 1.0 eq.) was dissolved in dichloromethane (100 mL) in a dry one-necked, 500 mL round bottom flask. Then 1 M hydrazine in THF (102.36 mL, 102.36 mmol, 1.5 eq.) was added dropwise. After stirring overnight at room temperature, the mixture was washed with sodium bicarbonate solution, 10% hydrochloric acid and sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The remaining liquid was purified by column chromatography (3:7 ethyl acetate/petroleum ether) to obtain an off-white oil (yield: 5.8 g, 24%).
1H NMR (300 Hz, 298 K, CDCl3, δ/ppm): 5.88–5.70 (m, 2H), 5.06–4.92 (m, 4H), 4.10–3.98 (m, 6H), 2.62–2.50 (m, 2H), 2.14–2.02 (m, 4H), 1.88–1.58 (m, 8H), 1.54–1.41 (m, 4H), 1.34 (t, J = 7.9 Hz, 1H).
31P{H} NMR (202 MHz, 298 K, CDCl3, δ/ppm): −0.68.
Poly-1
Poly-1 was prepared by a radical thiol–ene polyaddition. 33 g (94.2 mmol; 1.0 eq.) of the previously synthesized monomer 1 were dissolved in 230 mL toluene and added to a reactor fitted with a mechanical stirrer under an argon atmosphere. As a radical initiator, 1.4 g azobisisobutyronitrile (AIBN) (8.5 mmol; 0.1 eq.) was used. The solution was heated at 90 °C for 24 hours. The crude mixture was then concentrated and dried at reduced pressure until constant weight (yield: 32.5 g, 98.5%).1H NMR (300 Hz, 298 K, CDCl3, δ/ppm): 5.88–5.70 (m, 2H), 5.06–4.92 (m, 4H), 4.10–3.98 (m, 6H), 2.62–2.50 (m, 4H), 2.14–2.02 (m, 4H), 1.88–1.58 (m, 8H), 1.54–1.41 (m, 4H).
31P{H} NMR (202 MHz, 298 K, CDCl3, δ/ppm): −0.70.
Poly-2
The synthesis was done according to a literature procedure.11Poly-1 for kinetic studies
In a 25 mL Schlenk tube, 1 (405 mg, 1.16 mmol) was dissolved in toluene (2.7 mL) under an argon atmosphere. AIBN (0.03, 0.06, or 0.09 eq.) was added to the Schlenk tube and the mixture was heated to 70 °C or 90 °C. After specific reaction times, samples (each 0.2 mL) were taken and terminated in air for the analysis of the polymerization kinetics. The crude product was dried in vacuo and analyzed by 1H, 31P NMR, and GPC.1H NMR (300 Hz, 298 K, CDCl3, δ/ppm): 5.88–5.70 (m, 2H), 5.06–4.92 (m, 4H), 4.10–3.98 (m, 6H), 2.62–2.50 (m, 4H), 2.14–2.02 (m, 4H), 1.88–1.58 (m, 8H), 1.54–1.41 (m, 4H).
31P{H} NMR (202 MHz, 298 K, CDCl3, δ/ppm): −0.70.
Epoxy preparation
All epoxy resins were prepared using bisphenol A diglycidylether (DGEBA) (Araldite MY740, Bodo Möller Chemie GmbH, Offenbach am Main, Germany) as the epoxide agent and 2,2′-dimethyl-4,4′-methylene-bis-(cyclohexylamine) (DMC) (Sigma Aldrich Co. LLC/Merck KgaA, Darmstadt, Germany) as the amine hardener. The materials were mixed, poured into aluminum molds of desired dimensions, then hardened at 150 °C for 3 h. The flame retarded epoxy resins were produced in the same manner, except 10 wt% of the mixture was replaced with the respective flame retardant.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG WU 750/8-1; SCHA 730/15-1) for funding. Jens C. Markwart is the recipient of a fellowship through funding of the Excellence Initiative (DFG/GSC 266) in the context of the graduate school of excellence “MAINZ” (Materials Science in Mainz). F. R. Wurm and Jens C. Markwart thank Prof. Dr. Katharina Landfester (MPI-P, Germany) for support. Alexander Battig thanks Dr. Katharina Kebelmann and Patrick Klack for their support with Py-GC/MS and the cone calorimeter. Open Access funding provided by the Max Planck Society.References
- J. Liu, W. Huang, Y. Pang and D. Yan, Chem. Soc. Rev., 2015, 44, 3942–3953 RSC.
- M. D. Lechner, K. Gehrke and E. H. Nordmeier, Makromolekulare Chemie: Ein Lehrbuch für Chemiker, Physiker, Materialwissenschaftler und Verfahrenstechniker, Springer, Berlin Heidelberg, 2014 Search PubMed.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- B. Schartel and J. H. Wendorff, Polym. Eng. Sci., 1999, 39, 128–151 CrossRef CAS.
- K. Täuber, F. Marsico, F. R. Wurm and B. Schartel, Polym. Chem., 2014, 5, 7042–7053 RSC.
- B. Perret, B. Schartel, K. Stöβ, M. Ciesielski, J. Diederichs, M. Döring, J. Krämer and V. Altstädt, Eur. Polym. J., 2011, 47, 1081–1089 CrossRef CAS.
- B. Perret, B. Schartel, K. Stöβ, M. Ciesielski, J. Diederichs, M. Döring, J. Krämer and V. Altstädt, Macromol. Mater. Eng., 2011, 296, 14–30 CrossRef CAS.
- C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257–1281 CrossRef CAS.
- S. Penczek, K. Kaluzynski and J. Pretula, J. Appl. Polym. Sci., 2007, 105, 246–254 CrossRef CAS.
- J. Liu, W. Huang, Y. Pang, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2010, 11, 1564–1570 CrossRef CAS.
- A. Battig, J. C. Markwart, F. R. Wurm and B. Schartel, Polym. Chem., 2019, 10, 4346–4358 RSC.
- J. Khandare, M. Calderón, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- Y. Zhou, W. Huang, J. Liu, X. Zhu and D. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS PubMed.
- F. Marsico, A. Turshatov, R. Peköz, Y. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R. Wurm, J. Am. Chem. Soc., 2014, 136, 11057–11064 CrossRef CAS PubMed.
- M. M. Velencoso, A. Battig, J. C. Markwart, B. Schartel and F. R. Wurm, Angew. Chem., Int. Ed., 2018, 57, 17 CrossRef PubMed.
- P. Wen, X. Wang, W. Xing, X. Feng, B. Yu, Y. Shi, G. Tang, L. Song, Y. Hu and R. K. K. Yuen, Ind. Eng. Chem. Res., 2013, 52, 17015–17022 CrossRef CAS.
- J. Li, C. Ke, L. Xu and Y. Wang, Polym. Degrad. Stab., 2012, 97, 1107–1113 CrossRef CAS.
- K.-C. Cheng, C.-C. Wang, J.-l. Ruan, C.-H. Wu and C.-W. Li, Polym. Adv. Technol., 2018, 29, 2529–2536 CrossRef CAS.
- M. Rakotomalala, S. Wagner and M. Döring, Materials, 2010, 3, 4300 CrossRef CAS PubMed.
- K. H. Pawlowski and B. Schartel, Polym. Int., 2007, 56, 1404–1414 CrossRef CAS.
- B. Perret, K. H. Pawlowski and B. Schartel, J. Therm. Anal. Calorim., 2009, 97, 949 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1952, 74, 2718–2723 CrossRef CAS.
- D. Yan, C. Gao and H. Frey, Hyperbranched Polymers: Synthesis, Properties, and Applications, Wiley, 2011 Search PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- C. Walling, J. Am. Chem. Soc., 1945, 65, 441–447 CrossRef CAS.
- H. Tan, H. Liu, X. Chen, H. Chen and S. Qiu, Org. Biomol. Chem., 2015, 13, 9977–9983 RSC.
- J. C. Markwart and F. R. Wurm, Tetrahedron, 2018, 74, 7426–7430 CrossRef CAS.
- M. Ciesielski, A. Schäfer and M. Döring, Polym. Adv. Technol., 2008, 19, 507–515 CrossRef CAS.
- B. Schartel, C. A. Wilkie and G. Camino, J. Fire Sci., 2016, 34, 447–467 CrossRef.
- J. C. Markwart, A. Battig, L. Zimmermann, M. Wagner, J. Fischer, B. Schartel and F. R. Wurm, ACS Appl. Polym. Mater., 2019, 1, 1118–1128 CrossRef CAS.
- L.-H. Lee, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 859–882 CrossRef CAS.
- D. P. Bishop and D. A. Smith, J. Appl. Polym. Sci., 1970, 14, 205–223 CrossRef CAS.
- M. Ciesielski, B. Burk, C. Heinzmann and M. Döring, in Novel Fire Retardant Polymers and Composite Materials, ed. D.-Y. Wang, Woodhead Publishing, 2017, pp. 3–51, DOI:10.1016/B978-0-08-100136-3.00002-9.
- B. Schartel, B. Perret, B. Dittrich, M. Ciesielski, J. Krämer, P. Müller, V. Altstädt, L. Zang and M. Döring, Macromol. Mater. Eng., 2016, 301, 9–35 CrossRef CAS.
- B. Perret, K. Pawlowski and B. Schartel, J. Therm. Anal. Calorim., 2009, 97, 949–958 CrossRef CAS.
- S. Brehme, B. Schartel, J. Goebbels, O. Fischer, D. Pospiech, Y. Bykov and M. Döring, Polym. Degrad. Stab., 2011, 96, 875–884 CrossRef CAS.
- R. V. Petrella, J. Fire Sci., 1994, 12, 14–43 CrossRef CAS.
- B. Schartel and T. R. Hull, Fire Mater., 2007, 31, 327–354 CrossRef CAS.
- B. Schartel, M. Bartholmai and U. Knoll, Polym. Degrad. Stab., 2005, 88, 540–547 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C9PY01156K |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |